


Pulmonary endothelial heterogeneity shapes divergent molecular vulnerabilities under SMAD4 deficiency
 0
0 Abstract
Aim: Pulmonary vascular endothelial cells (ECs) comprise functionally distinct subpopulations, including arterial, venous, general capillary (gCap), and aerocyte capillary (aCap) ECs, that are essential for the maintenance of lung function. SMAD family member 4 (SMAD4) signaling is a critical contributor to vascular homeostasis. This study aims to elucidate how SMAD4 affects individual endothelial subpopulations in the lung.
Methods: We curated a previously published single-cell RNA sequencing (scRNA-seq) dataset from Smad4-deficient (Smad4fl/fl; Rosa26CreERT2) and control (Smad4fl/fl) mouse lungs, using a unified bioinformatic pipeline. Integrated analyses of cell clustering, differential gene expression, high-dimensional weighted gene co-expression network analysis (hdWGCNA), and chromatin immunoprecipitation sequencing were applied to resolve subpopulation-specific transcriptomic shifts and regulatory modules in the pulmonary endothelial compartment.
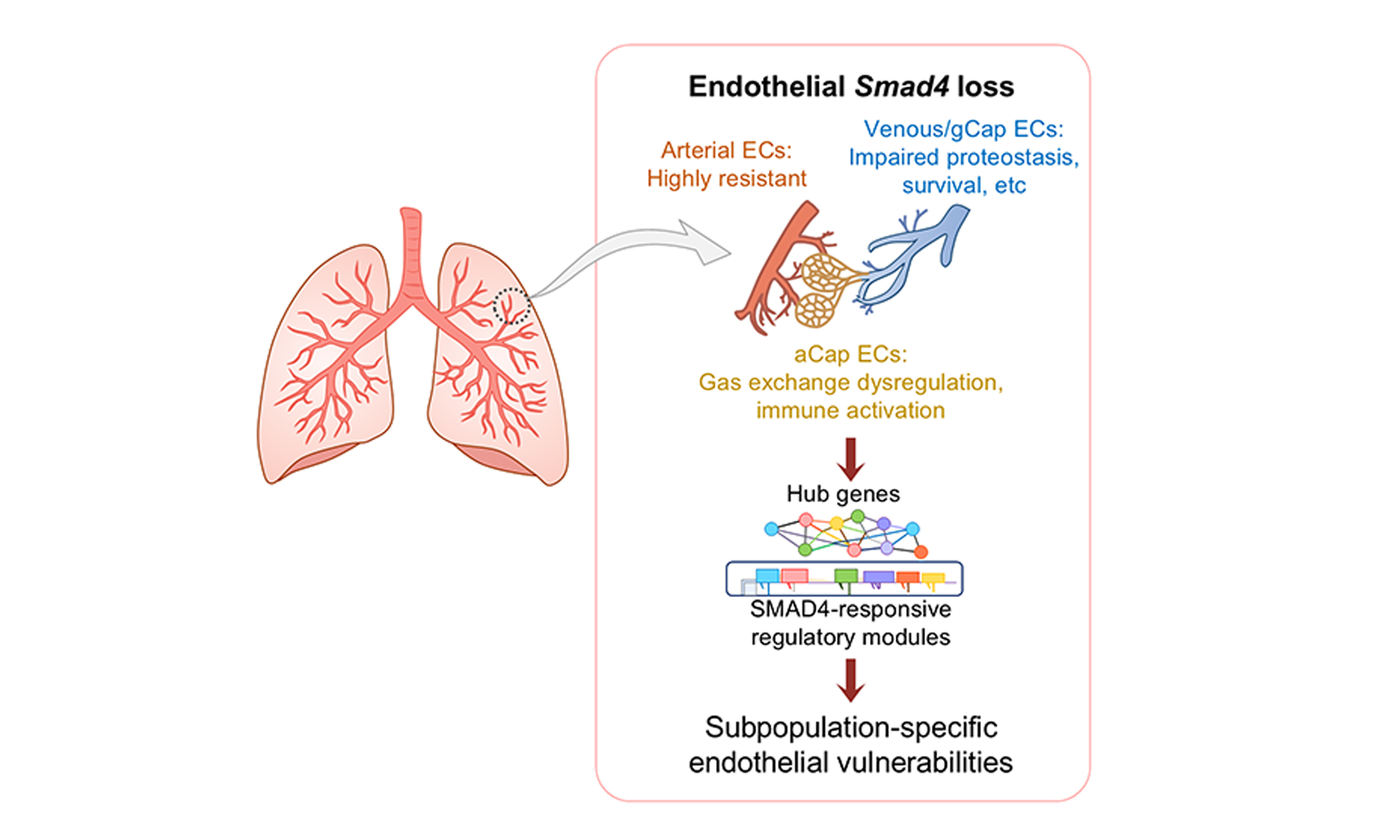
Results: SMAD4 deficiency induced markedly asymmetric transcriptomic changes across pulmonary vascular endothelial subpopulations. Upon Smad4 loss, arterial ECs largely preserved their core transcriptomic identity, whereas venous and gCap ECs transitioned into a vulnerable state characterized by aberrant proteostasis and reduced survival. Smad4-null gCap ECs also exhibited disrupted vascular homeostatic and morphogenic processes. Smad4-null aCap ECs displayed a distinct transcriptomic shift, manifested as gas-exchange dysregulation and immune activation, alongside shared defects in translation/protein folding and vascular morphogenic capacity. Regulatory modules and putative hub genes associated with venous/gCap EC vulnerability and aCap-specific transcriptomic shift upon SMAD4 deficiency were further identified.
Conclusion: These findings define a lineage-resolved framework for SMAD4 signaling in the pulmonary endothelium and uncover molecular vulnerabilities that may inform EC subpopulation-targeted vascular interventions.
Keywords
INTRODUCTION
The pulmonary vasculature plays a pivotal role in lung physiology. It is closely aligned with the alveolar-interstitial interface, enabling efficient ventilation-perfusion matching and gas exchange, and it also actively participates in immune regulation by facilitating pathogen sensing and leukocyte trafficking. In addition, the endothelial barrier preserves fluid balance and maintains interstitial pressure gradients essential for alveolar stability and respiratory mechanics[1].
Rather than functioning as a uniform population, pulmonary vascular endothelial cells (ECs) comprise functionally distinct subpopulations characterized by marked heterogeneity, including arterial, venous, general capillary (gCap), and aerocyte capillary (aCap) ECs[2]. These subpopulations form a spatially and functionally partitioned continuum, characterized by differences in cytoskeletal and extracellular matrix (ECM) organization, metabolic programming, and microenvironmental interactions. Consequently, a single upstream signaling input may elicit distinct transcriptional and functional outcomes across EC subpopulations. Understanding how lineage-specific specialization confers differential regulatory modes or vulnerabilities may lead to the discovery of specific vascular interventions for disease treatment.
SMAD family member 4 (SMAD4) is a central effector of the transforming growth factor-β (TGF-β)/bone morphogenetic protein (BMP) signaling pathways, transducing canonical signaling cascades downstream of the TGF-β receptor (TGFBR) or BMP receptor (BMPR) superfamily and thereby regulating a broad spectrum of cellular functions, such as cytoskeletal organization, mechanotransductive responses, metabolic balance, barrier function, and inflammatory regulation[3,4]. Genetic studies provide extensive evidence of its essential roles: global Smad4 deletion causes early embryonic lethality[5,6]; endothelial- or smooth-muscle-specific deletion disrupts vascular maturation and leads to arteriovenous malformations (AVMs)[7,8]; and inducible deletions of Smad4 in the endothelium in adulthood result in vascular instability, including impaired shear responsiveness, vascular leak and hemorrhagic phenotypes, and pulmonary hypertension (PH)[9-11]. However, most of these studies have treated the endothelium as a uniform compartment. It remains unclear whether SMAD4 influences pulmonary EC subpopulation in a similar or differential manner, or whether these EC lineages exhibit distinct susceptibilities in response to Smad4 loss.
In this study, we analyzed a previously published single-cell RNA sequencing (scRNA-seq) dataset derived from inducible, global Smad4-deficient mice and their controls. We identified arterial, venous, gCap, and aCap ECs and found that Smad4 loss triggered divergent changes in these vascular EC subpopulations. Arterial ECs remained largely stable, whereas venous and gCap EC lineages exhibited a conserved transcriptomic shift toward proteostasis suppression and impaired cell survival. aCap ECs displayed additional vulnerabilities associated with dysregulated gas exchange and immune activation. Integration of lineage-specific regulatory modules derived from high-dimensional weighted gene co-expression network analysis (hdWGCNA) and chromatin immunoprecipitation sequencing (ChIP-seq) revealed that specific vascular EC subpopulations engage distinct SMAD4-responsive regulatory gene sets. These findings provide mechanistic insights into EC lineage-specific vulnerabilities to Smad4 loss, which may underlie diverse forms of pulmonary vascular disease associated with SMAD4 deficiency.
METHODS
ScRNA-seq dataset acquisition
We reanalyzed a previously published scRNA-seq dataset[9] deposited in the Gene Expression Omnibus (GEO) database (GSE276257). This dataset was generated from seven batches of lung single-cell preparations obtained from 8-week-old mice, including three Smad4fl/fl controls and four inducible, global Smad4-null (Smad4fl/fl; Rosa26CreERT2, hereafter referred to as Smad4iko) mice that received five intraperitoneal
Data processing
Raw unique molecular identifier (UMI) count matrices downloaded from the GEO database were processed using Seurat v4.0.4. Quality control was performed on each sample prior to integration to remove low-quality cells. Cells were excluded if they had < 250 detected genes (nFeature_RNA), > 25% mitochondrial gene counts (percent.mt), > 5% hemoglobin/erythrocyte gene counts (percent.HB; computed from hydrogen bond acceptor/donor genes), or low molecular complexity (log10GenesPerUMI < 0.8). Rare genes detected in fewer than 10 cells and putative cell doublets identified by the scDblFinder function (DoubletScore < 10) were also removed.
After filtering, data were normalized using the SCTransform (SCT) function. For cross-sample integration, the top 3,000 variable genes in each sequencing sample were selected using SelectIntegrationFeatures on the normalized objects. Integration anchors across different sequencing samples were identified with FindIntegrationAnchors and used to generate an integrated expression matrix with IntegrateData, both using SCT-based integration. Following quality control and integration, 50,338 cells were retained for downstream single-cell analyses.
Cell clustering and annotation
To perform dimensionality reduction, the top 30 principal components were used to construct a k-nearest neighbor (KNN) graph. Cell clusters were identified using a Louvain graph-based clustering algorithm at standard resolution settings. Low-dimensional embeddings were visualized using uniform manifold approximation and projection (UMAP), and clusters were annotated based on canonical cell-type marker genes.
To resolve cellular heterogeneity, subclustering analysis of pan-endothelial populations (4,741 ECs in total) was performed at higher resolution to refine the boundaries of four vascular EC subpopulations (arterial, venous, gCap, and aCap ECs) and lymphatic ECs. Cluster-defining genes were identified using FindAllMarkers with the thresholding parameters: log2(fold change) > 0.25, adjusted p < 0.05, and min.pct>0.1. Differentially expressed genes (DEGs) between Smad4iko and Smad4fl/fl groups were identified using the FindMarkers function. Gene Ontology (GO) enrichment analysis and gene set enrichment analysis (GSEA) were also conducted, and statistical significance was determined as adjusted p or q values < 0.05 (Wilcoxon rank-sum test followed by Benjamini-Hochberg correction).
Principal component analysis
To assess whether Smad4 loss alters the transcriptomic state of arterial, venous, gCap, and aCap ECs, principal component analysis was performed on each subpopulation. Highly variable genes were selected for dimensionality reduction. The Seurat RunPCA function was applied to SCTransform residuals using the top 30 principal components, followed by the visualization of Smad4iko and Smad4fl/fl cells in principal component space to evaluate genotype-associated shifts.
hdWGCNA
hdWGCNA was used to identify lineage-specific regulatory networks associated with Smad4 loss. To reduce sparsity, transcriptionally similar cells within each endothelial lineage were grouped using KNNs and aggregated into metacells[12]. A k value of 10 was chosen to balance noise reduction and preservation of transcriptomic heterogeneity, and metacells were constructed to ensure robust estimation of gene-gene correlations[12]. Each metacell contained a minimum of 15 cells with homogeneous transcriptomic profiles. Mean expression values were used to generate metacell-level matrices, and genes detected in ≥ 10% of cells were retained for network construction. Following gene-gene correlation analysis at the metacell level, a weighted co-expression network was constructed using a soft-thresholding power of 4, approximating scale-free topology as recommended in the hdWGCNA framework[12]. A signed topological overlap matrix was calculated, and co-expression modules were identified by hierarchical clustering with dynamic tree cutting (DeepSplit = 4, detectCutHeight = 0.995, minClusterSize = 30, mergeCutHeight = 0.2), parameters commonly used to achieve fine-grained yet stable module detection in hdWGCNA workflows[12].
To identify genotype-responsive modules, we compared module eigengene distributions in the indicated EC subpopulations between Smad4iko and Smad4fl/fl genotypes. Modules whose eigengenes differed significantly between genotypes were defined as genotype-responsive. For each module, the top hub genes (top 10 ranked by the eigengene-based connectivity (kME)) were intersected with subpopulation-specific DEGs to define high-confidence regulatory candidates affected by Smad4 loss.
Re-analysis of ChIP-seq dataset
A SMAD4 ChIP-seq dataset generated from mouse lung tissue was retrieved from a publicly available database (GSE57177)[13]. BigWig locus tracks for ChIP and corresponding input control signals were visualized using Integrative Genomics Viewer (IGV) software (v2.16.0) on the mm9 genome assembly. Genomic coordinates were used to inspect promoter-proximal regions (covering the region from -5 kb to
Software and statistics
All scRNA-seq analyses were performed using the R software (v4.1.0). ChIP-seq signal visualization was conducted using the IGV software (v2.16.0). The Wilcoxon rank-sum test followed by Benjamini-Hochberg correction was used for statistical analysis of the transcriptomic data, with adjusted p or q values < 0.05 considered statistically significant.
RESULTS
Single-cell atlas of vascular EC subpopulations in control and Smad4-deficient mouse lungs
To investigate the transcriptomic signatures of pulmonary vascular EC subpopulations, we integrated our previously published scRNA-seq dataset[9] from 8-week-old control (Smad4fl/fl) and inducible, global Smad4-null (Smad4iko) mice, generating a comprehensive atlas encompassing all major pulmonary cell types (ECs, fibroblasts, airway smooth muscle cells (ASMCs), vascular smooth muscle cells (VSMCs), monocytes/macrophages, dendritic cells, neutrophils, B cells, natural killer (NK)/T cells, and epithelial cells; Figure 1A).
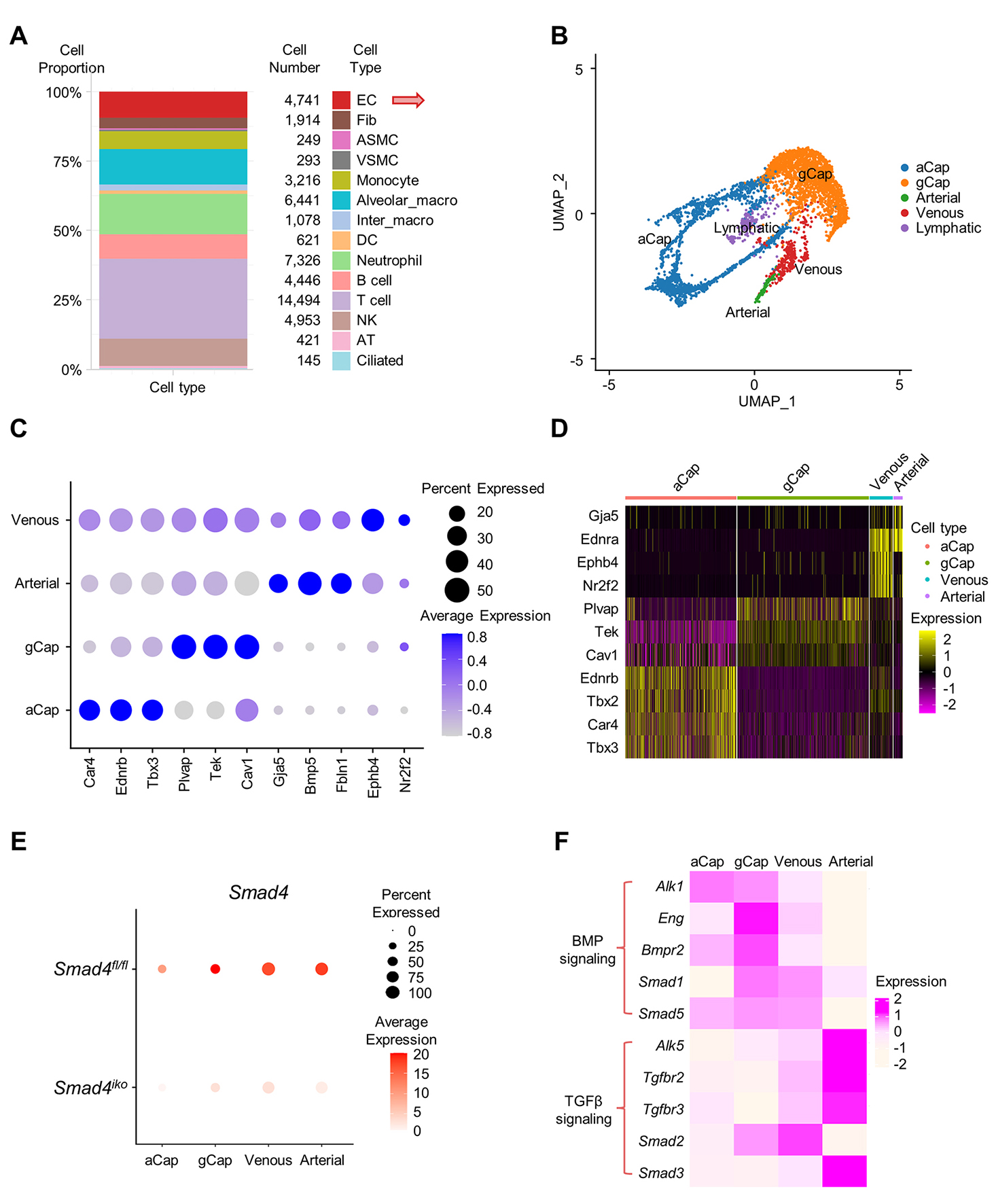
Figure 1. Single-cell transcriptomic profiling showing four pulmonary vascular EC subpopulations. (A) Proportion and numbers of lung cell populations identified in this study. (B) Uniform manifold approximation and projection (UMAP) visualization of pulmonary vascular EC subpopulations, including arterial, venous, general capillary (gCap) and aerocyte capillary (aCap) ECs, as well as lymphatic ECs. (C) Dot plot displaying expression pattern of canonical EC subtype marker genes (Car4, Ednrb, Tbx3, Plvap, Tek, Cav1, Gja5, Bmp5, Fbln1, Ephb4 and Nr2f2) across the identified vascular EC subpopulations. Dot size and color scale reflect the frequency of cells in the indicated clusters expressing the marker genes and average log-transformed expression intensity, respectively. (D) Heatmap showing expression profile of classical EC markers in the EC subpopulations in (C) Shown are arterial EC markers (Gja5, Ednra), venous EC markers (Nr2f2, Ephb4), capillary-associated genes (Plvap, Car4), and additional markers (Tek, Cav1, Ednrb, Tbx2, Tbx3). Distinct expression boundaries indicate accurate annotation of each EC lineage. (E) Dot plot showing Smad4 expression across pulmonary vascular EC subpopulations in Smad4fl/fl and Smad4iko mice. Dot size indicates the percentage of cells expressing Smad4, and dot color represents the average expression levels of Smad4. Note that Smad4 is broadly expressed in the four vascular EC subpopulations, and that Smad4 expression is markedly reduced in all subpopulations in Smad4iko mice compared with Smad4fl/fl controls, attesting to successful gene deletion. (F) Heatmap showing expression pattern of the indicated signaling components of BMP/ALK1 and TGF-β/ALK5 pathways across pulmonary vascular EC subpopulations. Color scale represents average log-transformed expression intensity. EC: Endothelial cell; Fib: fibroblast; VSMC: vascular smooth muscle cell; ASMC: airway smooth muscle cell; Alveolar_macro: alveolar macrophage; Inter_macro: interstitial macrophage; DC: dendritic cell; NK: natural killer cell; AT: alveolar epithelial cell.
The previous study treated all vascular ECs as a single compartment[9]. To examine subpopulation-specific effects of Smad4 loss, we performed subclustering analyses on the pan-EC population. Single-cell transcriptomes from 4,741 pulmonary ECs were obtained for downstream analysis. UMAP resolved five transcriptionally discrete EC lineages (arterial, venous, gCap, aCap, and lymphatic ECs, Figure 1B), with each subpopulation ranging from 150 to 2,156 cells and the number of detected genes ranging from 14,189 to 16,134 [Supplementary Table 1]. We then focused on the four vascular EC subpopulations. Canonical markers, including Car4/Ednrb/Tbx3 for aCap ECs, Plvap/Tek/Cav1 for gCap ECs, Gja5/Bmp5/Fbln1 for arterial ECs, and Ephb4/Nr2f2 for venous ECs, confirmed the accuracy of subpopulation annotation
SMAD4 acts as a common effector of TGF-β and BMP signaling pathways. Analysis of the expression patterns of TGFBR and BMPR and downstream SMADs suggested enriched expression of TGF-β/activin receptor-like kinase 5 (ALK5) signaling components in arterial ECs compared to the other three EC subpopulations, in contrast to the two capillary EC subpopulations in which BMP/activin receptor-like kinase 1 (ALK1) signaling components are preferentially expressed [Figure 1F]. Venous ECs did not display a clearly segregated expression pattern between the TGF-β and BMP signaling axes [Figure 1F]. The differences in the basal expression levels of these signaling components raise the possibility that lung EC subpopulations have distinct reliance on the ALK1 versus ALK5 axes, which may give rise to subpopulation-specific transcriptomic responses to upstream signaling inputs.
Arterial ECs undergo minimal transcriptomic changes after Smad4 loss
To assess the impact of SMAD4 deficiency on arterial ECs, we performed principal component analysis of scRNA-seq profiles from Smad4iko and Smad4fl/fl genotypes. Smad4iko and Smad4fl/fl arterial ECs largely overlapped in the principal component space without obvious genotype-associated separation [Figure 2A], indicating limited divergence of the overall transcriptomic state. In line with this, only a small subset of DEGs was identified in arterial ECs between the genotypes [Figure 2B]. Functional enrichment analyses revealed only a few enriched pathways, primarily related to the regulation of autophagy and immune response-associated processes [Figure 2C and D]. GSEA also detected no significant changes in the expression levels of gene sets linked to core arterial functions, including vascular morphogenesis, hemodynamic responses, and endothelial barrier-associated pathways [Figure 2D]. Together, these results indicate that following Smad4 loss, arterial ECs underwent very limited changes in immune response-related pathways and largely preserved their core transcriptomic identity, despite their enriched expression of TGF-β/ALK5 signaling components.
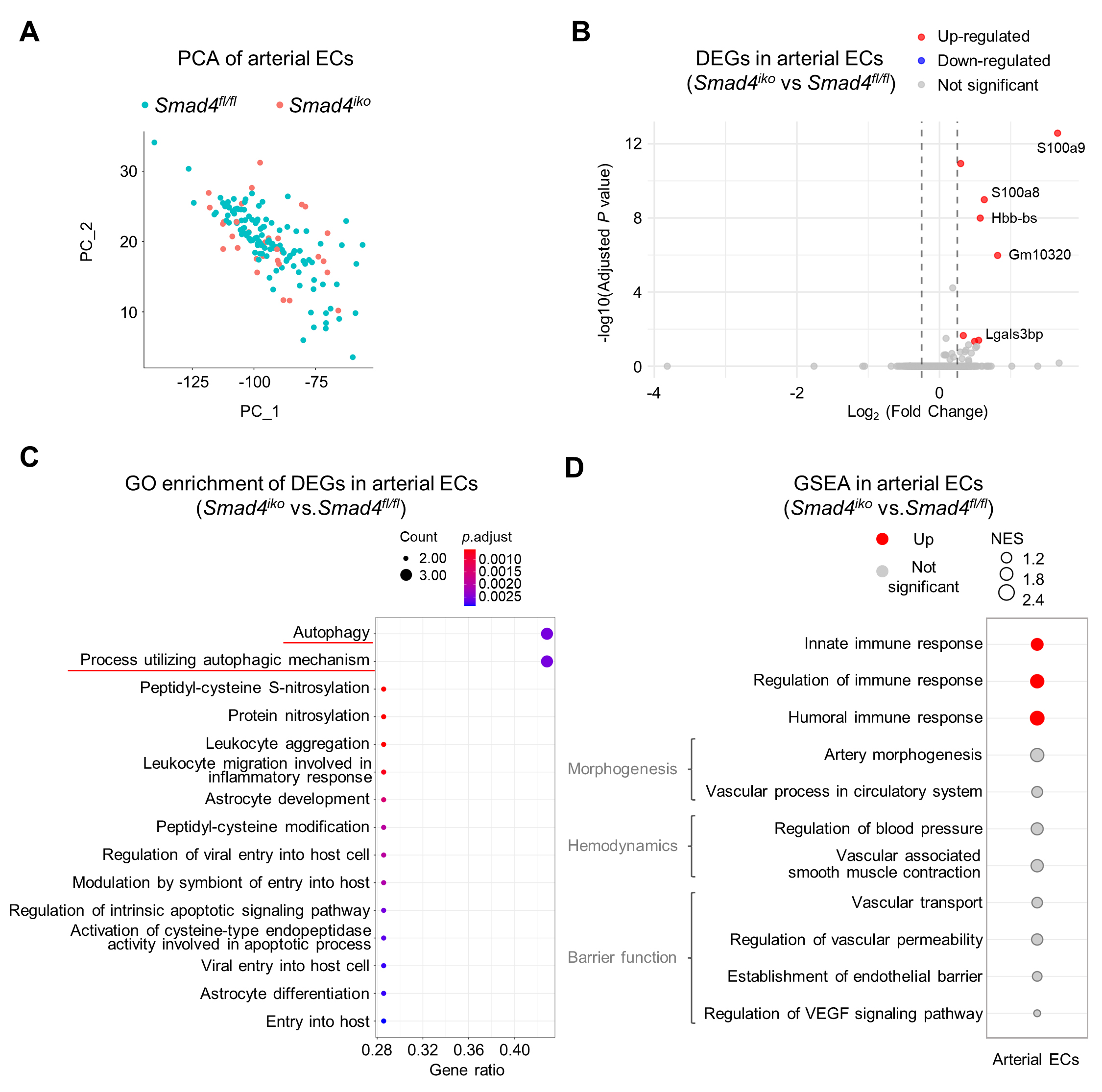
Figure 2. Transcriptomic profiling showing minimal alterations in pulmonary arterial ECs upon Smad4 loss. (A) Principal component analysis (PCA) of pulmonary arterial ECs between Smad4fl/fl and Smad4iko genotypes. Note largely overlapping distribution of ECs with the two genotypes, indicating their transcriptomic similarity. (B) Volcano plot analysis of differentially expressed genes (DEGs) in Smad4iko vs. Smad4fl/fl pulmonary arterial ECs. Upregulated and downregulated genes are labeled in red and blue, respectively. Note that only a few genes are modestly upregulated (see gene symbols). (C) Gene Ontology (GO) enrichment analysis of DEGs from arterial ECs. Note that only a small subset of biological processes displayed mild enrichment, including autophagy and immune response pathways (red lines). (D) Gene set enrichment analysis (GSEA) showing the expression profile of the indicated functional processes in Smad4iko vs. Smad4fl/fl pulmonary arterial ECs. NES: normalized enrichment score.
Characterization of common and lineage-specific transcriptomic signatures in venous and gCap ECs upon Smad4 loss
Compared with arterial ECs, venous and gCap ECs are exposed to relatively low pressure and shear stress, rendering them inherently more sensitive to genetic or microenvironmental perturbations[14,15]. Accordingly, we performed principal component analysis on scRNA-seq profiles of venous and gCap ECs from Smad4iko and Smad4fl/fl mice to evaluate the transcriptomic impact of Smad4 deficiency. Venous and gCap ECs from the two genotypes displayed clear separation in principal component space [Figure 3A and B], indicating genotype-associated transcriptomic differences. Indeed, DEG analysis revealed a substantial fraction of genes was significantly upregulated or downregulated in venous and gCap ECs between Smad4iko and Smad4fl/fl genotypes [Figure 3C and D]. GO enrichment analysis highlighted that a number of protein folding-related and apoptosis-related pathways were significantly altered both in venous and gCap ECs following Smad4 deletion [Figure 3E and F]. GSEA further revealed coordinated downregulation of apoptotic and protein folding-related pathways in both venous ECs and gCap ECs between Smad4iko and Smad4fl/fl genotypes [Figure 3G]. These data indicate a common role of SMAD4 signaling in the maintenance of proteostasis and appropriate survival activity in these EC subpopulations.
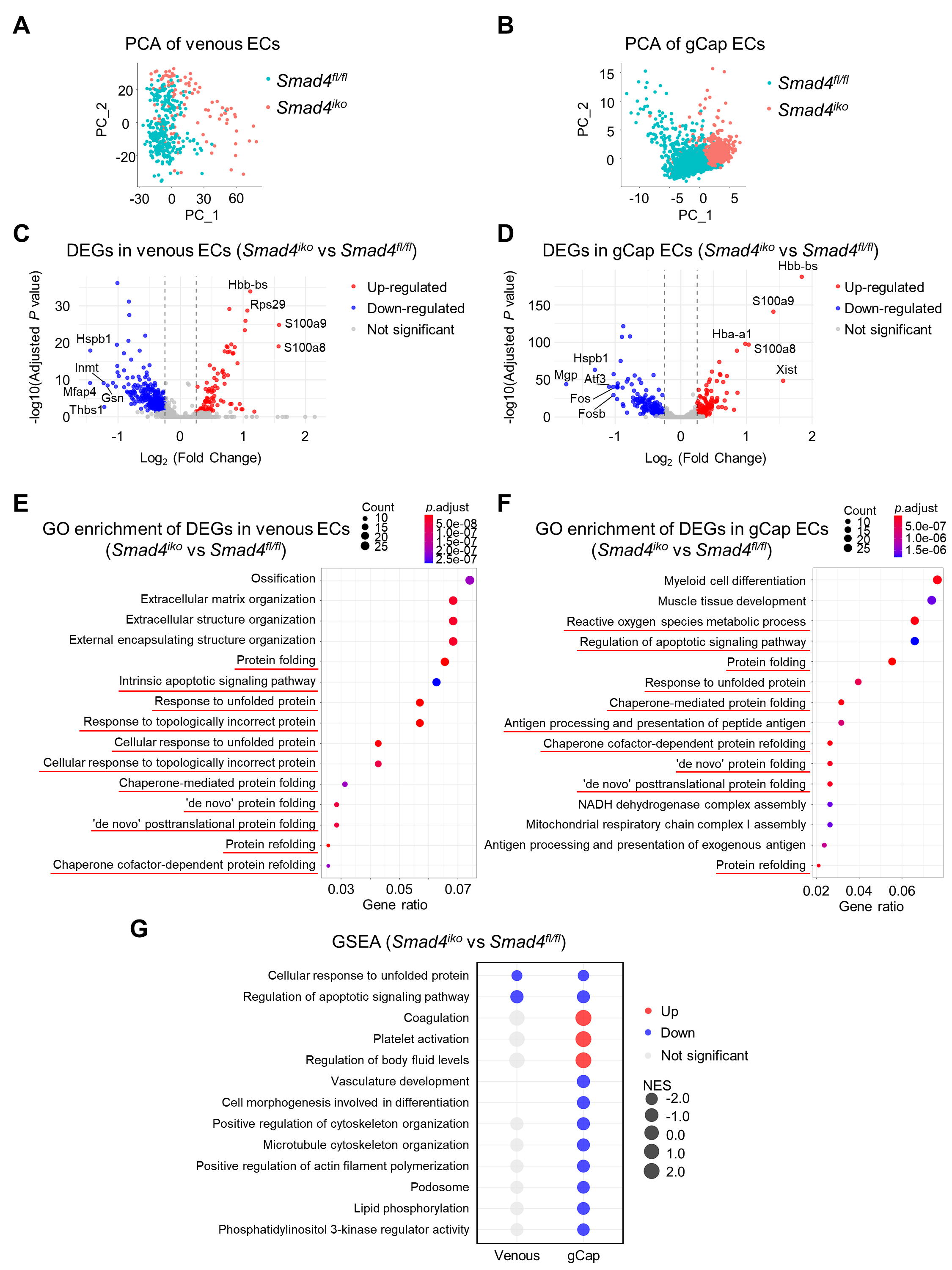
Figure 3. Characterization of the impact of Smad4 deletion on single-cell transcriptomics of pulmonary venous and gCap ECs. (A and B) Principal component analysis (PCA) of pulmonary venous ECs (A) and gCap ECs (B) from Smad4fl/fl and Smad4iko mice. Note that ECs from the two genotypes show a clear separation in both venous and gCap compartments, indicating genotype-associated transcriptomic differences. (C and D) Volcano plots of DEGs in Smad4iko vs. Smad4fl/fl pulmonary venous and gCap ECs. Significantly upregulated and downregulated genes are labeled in red and blue. Gene symbols indicate the top DEGs. (E and F) GO enrichment analysis of DEGs from pulmonary venous and gCap ECs. Note that a number of protein folding-related processes and apoptotic pathways are enriched in both EC subpopulations (red lines), indicating a common effect of SMAD4 on the maintenance of proteostasis and survival in these cells. (G) GSEA showing the indicated upregulated and downregulated pathways in Smad4iko vs. Smad4fl/fl pulmonary venous and gCap ECs. NES: normalized enrichment score. Note that Smad4iko gCap ECs show additionally upregulated pathways involved in coagulation, platelet activation and fluid homeostasis but downregulated pathways associated with vascular morphogenesis, cell morphology and cytoskeletal regulation, and phospholipid/PI3K signaling.
Apart from these shared transcriptomic changes, Smad4 loss exerted additional effects on gCap ECs compared with venous ECs, as evidenced by a broader set of upregulated and downregulated pathways [Figure 3G]. For example, gene sets related to vascular homeostatic pathways, such as coagulation, platelet activation, and body fluid retention, were significantly upregulated in Smad4iko versus Smad4fl/fl gCap ECs but not venous ECs [Figure 3G]. Conversely, Smad4iko led to significant downregulation of pathways linked to vascular or cell morphogenesis and associated cytoskeletal and membrane remodeling processes, including microtubule and actin cytoskeleton, podosome, and phosphoinositide 3-kinase (PI3K)-phospholipid signaling [Figure 3G].
Collectively, these findings indicate that venous and gCap ECs share convergent transcriptomic changes following SMAD4 deficiency in terms of proteostasis and survival, whereas gCap ECs exhibit a broader spectrum of vulnerabilities in vascular homeostatic and morphogenic processes in response to Smad4 loss.
Smad4 loss leads to a distinct transcriptomic shift in aCap ECs toward gas-exchange dysregulation and immune activation
Principal component analysis further revealed a clear separation in the principal component space between Smad4iko and Smad4fl/fl aCap ECs, indicating a Smad4 loss-associated transcriptomic shift [Figure 4A]. DEG analysis confirmed marked changes in the aCap EC subpopulation following Smad4 deletion, as evidenced by a substantial set of upregulated and downregulated genes [Figure 4B]. GO enrichment showed that these DEGs were enriched in pathways involved in ribonucleoprotein complex assembly, ribosome biogenesis, protein folding, and vascular structure development [Figure 4C]. GSEA further demonstrated significant downregulation of gene sets related to protein folding, translational control, and ribosomal structural components in Smad4iko aCap ECs [Figure 4D], in a similar trend to those observed in venous and gCap ECs. These data reinforce the notion that SMAD4 acts as a key regulator of proteostasis and vascular morphogenesis.
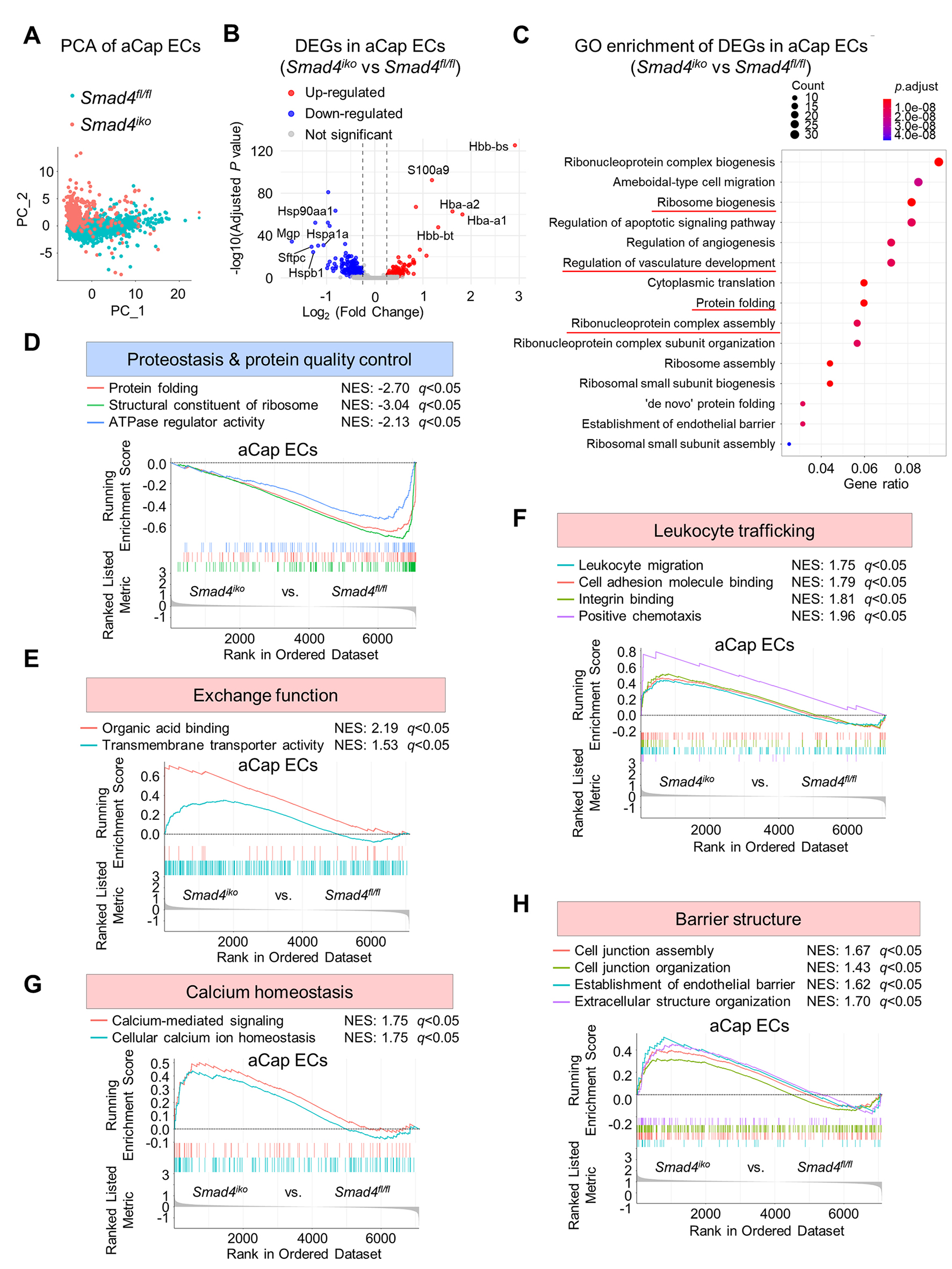
Figure 4. Transcriptomic shift in pulmonary aCap ECs induced by Smad4 loss, featuring dysregulated exchange function and activated immune responses. (A) Principal component analysis (PCA) of pulmonary aCap ECs from Smad4fl/fl and Smad4iko mice. Note that the two genotypes are separated, suggesting genotype-dependent transcriptomic alterations in aCap ECs. (B) Volcano plots of DEGs in Smad4iko vs. Smad4fl/fl pulmonary aCap ECs. Significantly upregulated and downregulated genes are labeled in red and blue. (C) GO enrichment analysis of DEGs from pulmonary aCap ECs. Note that a number of protein folding-related processes and apoptotic pathways are significantly enriched (red lines), reinforcing the notion that SMAD4 maintains proteostasis in ECs. (D) GSEA showing downregulated pathways mapped to proteostasis in Smad4iko vs. Smad4fl/fl pulmonary aCap ECs. NES: normalized enrichment score. (E-H) GSEA showing the indicated upregulated pathways linked to exchange function, leukocyte trafficking, calcium homeostasis and barrier architecture regulation in Smad4iko vs. Smad4fl/fl pulmonary aCap ECs.
aCap ECs are key components of the gas-exchange interface and enriched for leukocyte adhesion and immune cell-trafficking regulators, reflecting their dual roles in the alveolar-capillary gas-exchange interface and immunologic communication[15-18]. Smad4 deletion markedly disrupted these lineage-specific functions, producing a transcriptional shift distinct from that in arterial, venous, or gCap ECs. With respect to gas-exchange function, Smad4 loss significantly upregulated transmembrane transport-related pathways in aCap ECs, indicating increased exchange activity at the alveolar-capillary interface [Figure 4E]. In parallel, GSEA revealed broad upregulation of inflammatory pathways in Smad4iko aCap ECs, including leukocyte migration, leukocyte adhesion, chemotaxis, and integrin-mediated adhesive interactions, suggesting a transition toward an immune-activated state upon SMAD4 deficiency [Figure 4F]. In addition, gene sets mapped to calcium homeostasis, calcium signaling, and organic acid binding were also upregulated, supporting a role for SMAD4 in maintaining ionic balance at the alveolar-capillary interface
hdWGCNA prioritizes co-expression modules and hub genes in Smad4-null venous/gCap and aCap ECs
Building on the lineage-specific transcriptomic responses as described above (see Figures 2-4), we applied hdWGCNA to identify co-expression modules and prioritize hub gene candidates within EC subpopulations associated with Smad4 loss. As established above, the four pulmonary vascular EC subpopulations differ markedly in their response magnitudes following Smad4 loss. Arterial ECs had overall minimal transcriptomic changes, and we treated arterial ECs as a transcriptomically stable subpopulation resistant to SMAD4 deficiency and excluded them from hdWGCNA. Venous and gCap ECs exhibited a shared pattern of weakened proteostasis and survival, and aCap ECs displayed a distinct transcriptional shift featuring gas-exchange dysregulation and immune activation. Therefore, we conducted a unified analysis after combining venous and gCap ECs, whereas aCap ECs were analyzed independently.
hdWGCNA on the combined venous/gCap EC subpopulations identified seven co-expression modules, as shown by the gene cluster dendrogram and corresponding module gene counts [Figure 5A and B]. Among these, five modules were significantly altered in Smad4iko mutants, underscoring their association with SMAD4 deficiency [Figure 5C]. GO enrichment revealed that these significantly altered modules predominantly mapped to protein folding processes and apoptosis programs [Supplementary Figure 1A], largely recapitulating the results shown in Figure 3. To pinpoint key regulatory nodes, we collected the top 10 kME-ranked genes in each significant module (50 genes in total) and intersected them with venous/gCap DEGs, yielding 14 high-confidence hub genes [Figure 5D]. These hub genes were primarily heat-shock proteins (Hsp family members), stress-responsive transcription factors (e.g., Atf3), and ribosomal proteins (Rps/Rpl) [Figure 5E], raising the possibility that these hub genes might be responsible for the shared vulnerabilities in Smad4iko venous/gCap ECs to proteostasis and stress susceptibility.
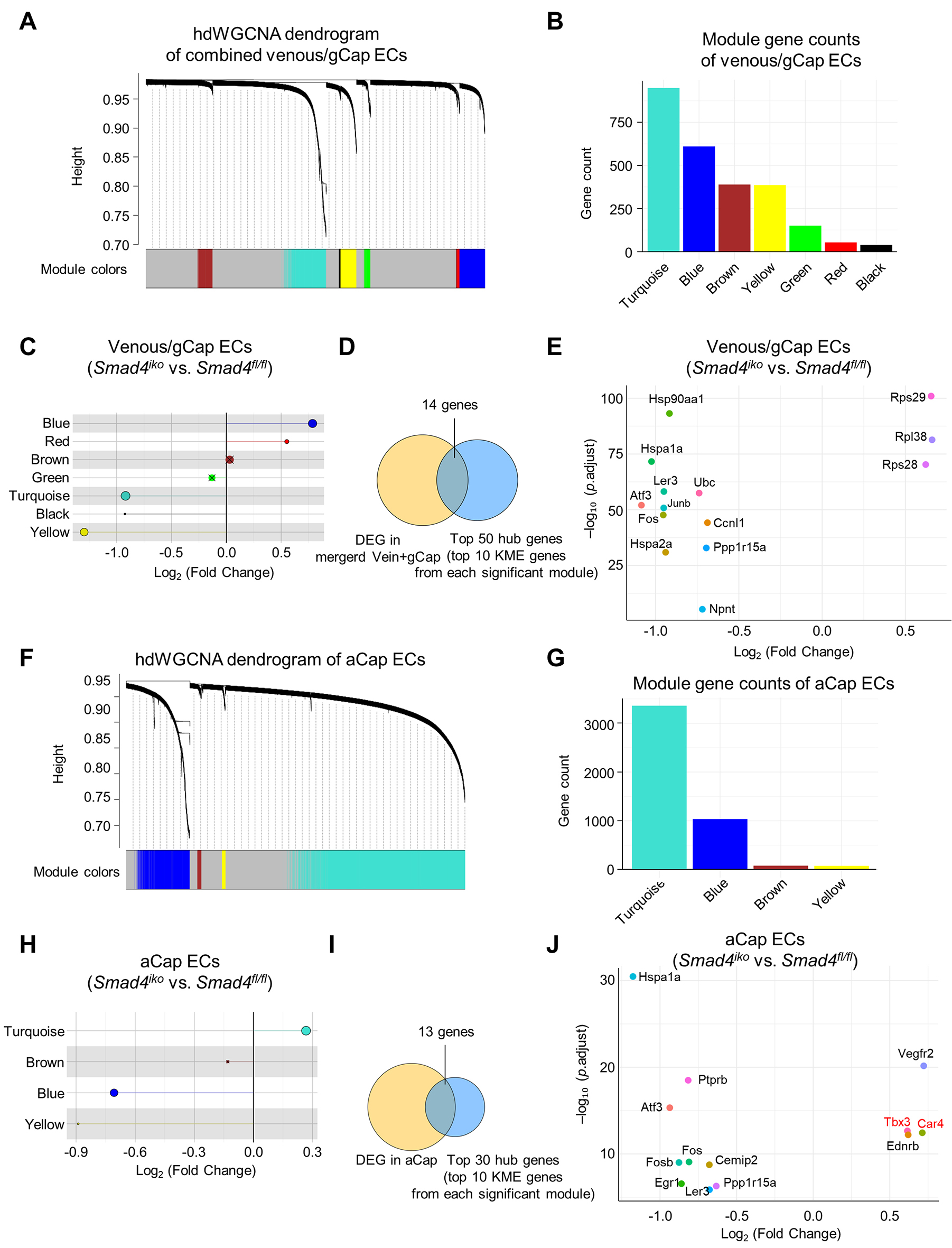
Figure 5. hdWGCNA identification of co-expression modules and hub genes within Smad4-null venous/gCap and aCap ECs. (A and F) Gene cluster dendrogram with color module assignments, as determined by high-dimensional weighted gene co-expression network analysis (hdWGCNA) of pulmonary venous/gCap (A) or aCap ECs (F) from both Smad4fl/fl and Smad4iko mice based on a Metacell-based approach. (B and G) Number of genes in co-expression modules as identified in (A) or (F). (C and H) Lollipop plot showing differentially expressed modules in Smad4fl/fl and Smad4iko pulmonary venous/gCap (C) or aCap ECs (H), as determined by module eigengene analysis. Crosses (X) indicate that the modules are not significantly expressed. (D and I) Venn diagram showing the intersection between DEGs and top hub-gene candidates (top 10 kME genes per significant module) in pulmonary venous/gCap (D) or aCap ECs (I), yielding 14 or 13 high-confidence hub genes, respectively. (E and J) Dot plot showing expression pattern of the high-confidence hub genes, as identified in (D) or (I).
To dissect aCap-specific co-expression modules, we performed an independent hdWGCNA on aCap ECs, which yielded a gene cluster dendrogram and module gene count distributions [Figure 5F-G], identifying three co-expression modules that were significantly associated with the Smad4iko genotype [Figure 5H]. Functional annotation revealed that these modules were related to cell-cell junctions, leukocyte migration and adhesion, gas exchange, cytoskeletal dynamics, and energy metabolism [Supplementary Figure 1B], consistent with the observations shown in Figure 4. By intersecting the top 10 kME-ranked genes from each module (30 genes in total) with aCap DEGs, we identified 13 genes, including Car4, Tbx3, and Ednrb, which are canonical aCap EC markers and key regulators of gas exchange and microcirculatory function and were upregulated in Smad4-null aCap ECs, and Egr1, an inflammatory regulator that was downregulated in Smad4-null aCap ECs [Figure 5I-J]. These genes are the putative hub genes that may drive Smad4 loss-associated abnormalities in aCap ECs. Together, hdWGCNA delineates the regulatory modules and putative hub genes that contribute to venous/gCap EC vulnerability and aCap-specific transcriptomic shift, providing co-expression-based evidence for EC subpopulation-specific signatures associated with SMAD4 deficiency.
Finally, to evaluate the regulatory relationship between SMAD4 and these hub genes, we re-analyzed a publicly available SMAD4 ChIP-seq dataset generated from mouse lung tissue. Among the 22 hub genes, including 5 genes that were common hub genes identified in both venous/gCap and aCap EC subpopulations, we observed SMAD4 binding peaks at the promoter-proximal regions centered on the TSS of 13 genes, including Atf3, Tbx3, Car4, Ccnl1, Egr1, Fos, Fosb, Hsp90aa1, Hspa2a, Ier3, Rpl38, Rps29 and Ubc [Figure 6 and Supplementary Figure 2]. These binding events support direct transcriptional regulation by SMAD4, whereas hubs lacking evident peaks likely represent indirect SMAD4-responsive genes.
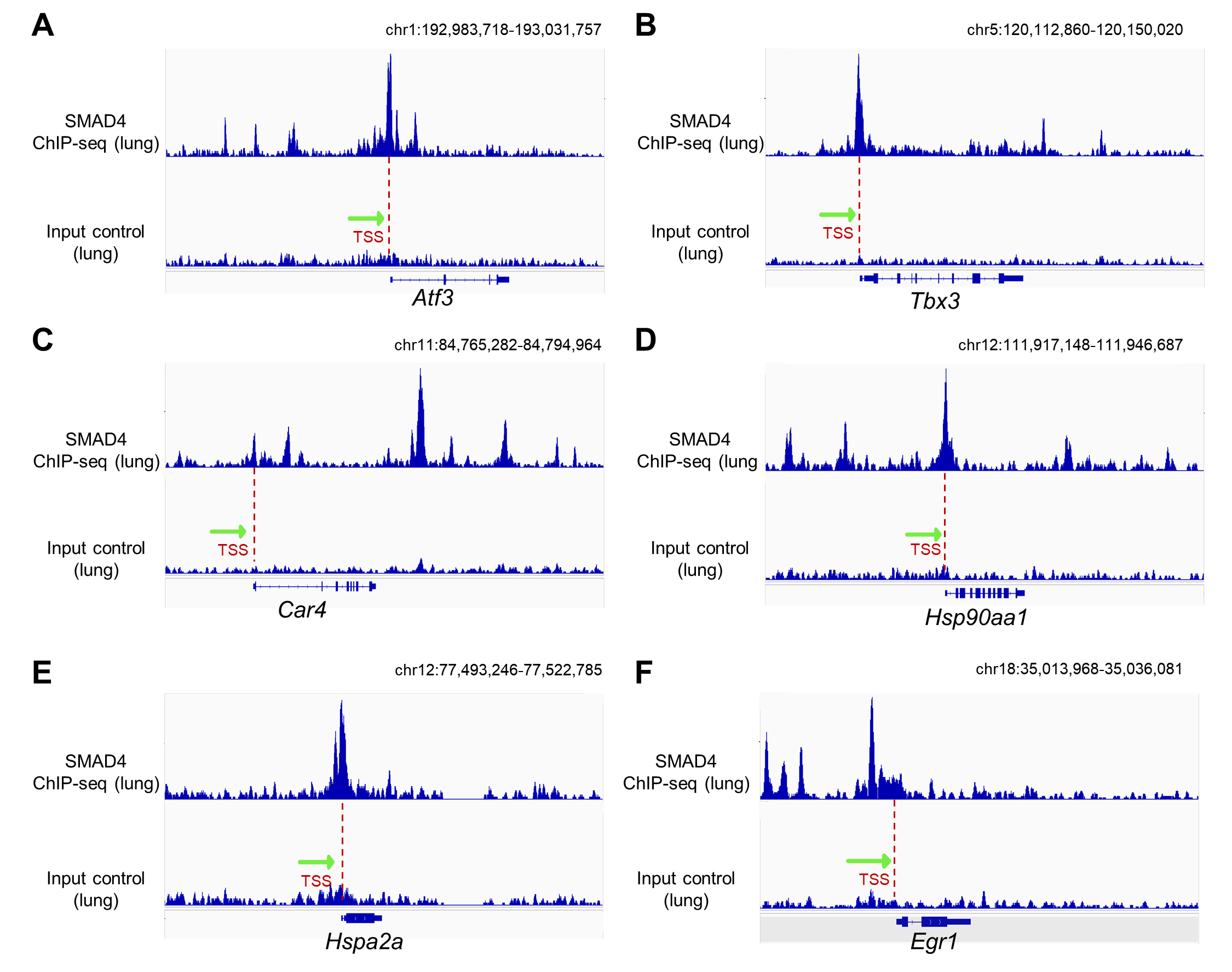
Figure 6. ChIP-seq analysis of potential SMAD4 target genes. (A-F) Bigwig locus tracks of SMAD4 ChIP (top panels) and corresponding input control signals (bottom panels) obtained from mouse lung tissue. SMAD4 binding peaks can be visualized around the transcription start site (TSS) of the Atf3 (A), Tbx3 (B), Car4 (C), Hsp90aa1 (D), Hspa2a (E) and Egr1 (F) locus. Red dashed lines indicate TSSs of the respective gene loci, and green arrows denote the direction of transcription.
DISCUSSION
SMAD4 signaling critically regulates vascular homeostasis. To date, insights into SMAD4 function have been derived primarily from organism- or tissue-level vascular phenotypes, such as aberrant angiogenesis, vascular malformations, and endothelial barrier dysfunction[19,20]. These studies often conceptualize vascular ECs as a single compartment, despite evidence for pronounced intrinsic heterogeneity of the vascular endothelium across organs, including the lung[21]. In this study, we show lineage-specific transcriptomic changes in pulmonary vascular EC subpopulations induced by SMAD4 deficiency. Our results, as summarized in Figure 7, reveal that arterial ECs are relatively resistant to SMAD4 deficiency, whereas Smad4 loss in venous and gCap ECs commonly perturbs pathways linked to proteostasis and survival, and aCap ECs undergo a distinct transcriptomic shift toward gas-exchange dysregulation and immune activation. These findings highlight that endothelial heterogeneity shapes divergent molecular vulnerabilities and may inform endothelial subpopulation-targeted vascular interventions.
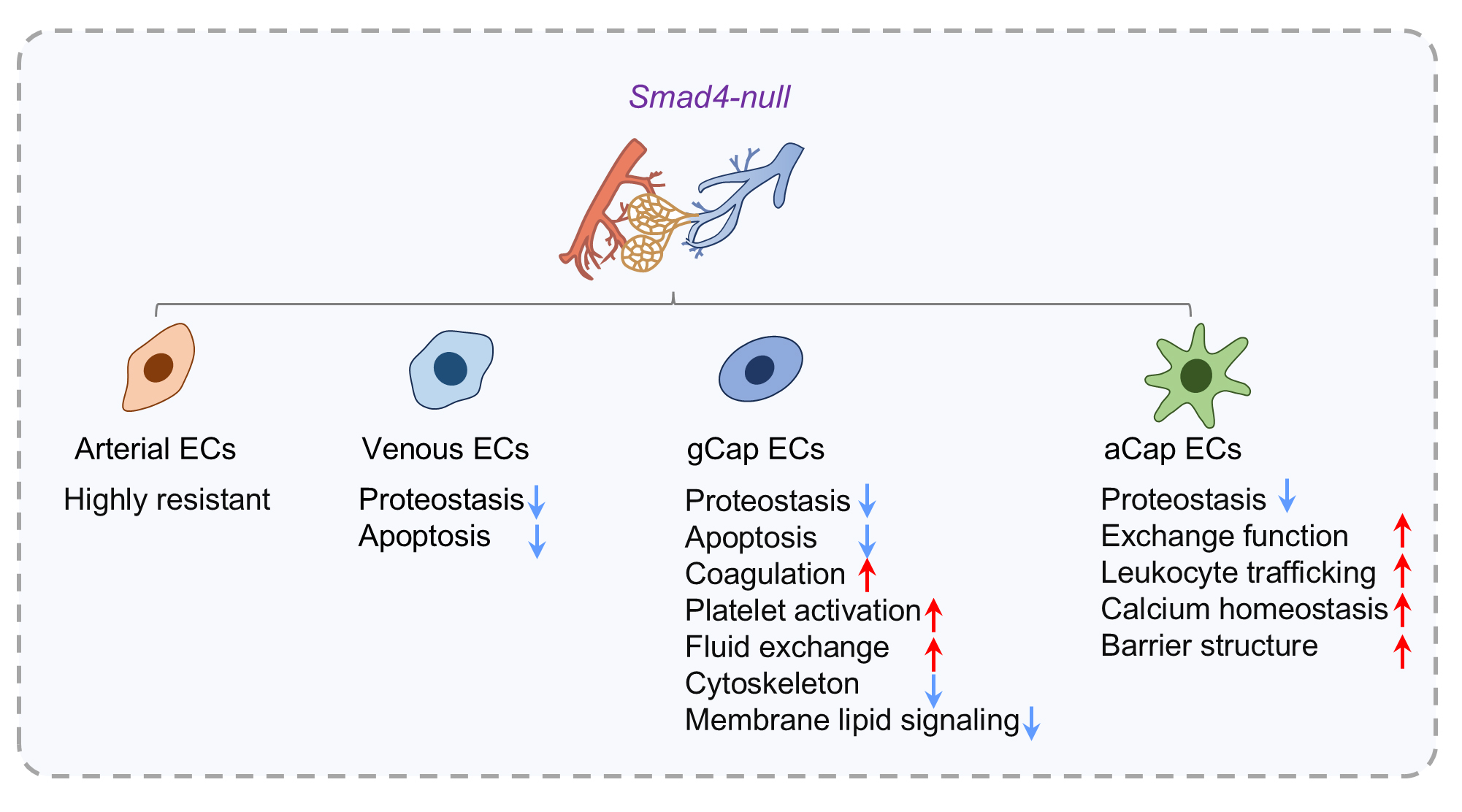
Figure 7. Summary model illustrating the impact of Smad4 loss on pulmonary vascular EC subpopulations.
The canonical BMP signal typically activates SMAD1/5/8, and TGF-β predominantly activates SMAD2/3[22]. Importantly, in ECs these two signaling inputs are largely segregated by specific receptors. ALK1 preferentially transduces BMP9/10-driven SMAD1/5/8 signaling, whereas ALK5 primarily mediates the TGF-β-SMAD2/3 axis[23]. These signaling cascades cooperatively shape endothelial homeostasis by critically regulating EC migration and proliferation as well as vascular maturation and stabilization[24]. Despite the common requirement for SMAD4 as the common SMAD to enable nuclear transcriptional output, lung EC subpopulations display differential expression patterns of the upstream receptors and downstream receptor-regulated SMADs (R-SMADs). As such, SMAD4 function is likely contingent on context-specific signaling networks in these EC subpopulations, and the potentially differential reliance on BMP/ALK1 versus TGF-β/ALK5 axes may account for the subpopulation-specific transcriptomic alterations upon Smad4 loss.
The transcriptome of arterial ECs showed strong resistance to Smad4 loss, with only very limited perturbations. A possible explanation is that arterial ECs are continuously exposed to a harsh mechanical environment characterized by high shear, pulsatile pressure, and transmural gradients, which requires transcriptomic stability to sustain strict coupling among cell-ECM adhesions, cytoskeletal tension, and junctional architecture in the artery[25,26]. In contrast to the relative stable arterial ECs, the pronounced transcriptional changes in venous and gCap ECs following Smad4 loss may reflect their high plasticity and remodeling capacity[27,28]. Our prior work[9] demonstrated that endothelial Smad4 loss promotes PH development. However, SMAD4 mutations are rare in human pulmonary arterial hypertension (PAH)[29]. Only ECs located in the core of the plexiform lesions lacked expression of SMAD4, whereas SMAD4 expression was preserved in ECs in the segment that feeds the lesion and in ECs of larger pulmonary arteries[30]. When comparing disease phenotypes, we noted that the PH manifestations associated with endothelial Smad4 loss are not fully identical to the PAH phenotype caused by BMPR2 (bone morphogenetic protein receptor type II) mutations, suggesting that Smad4 deficiency may drive pulmonary vascular remodeling through distinct mechanisms. Based on this divergence, we previously speculated that Smad4 loss-associated PH is more likely secondary to vascular malformations or hemorrhage-related lesions. In this context, hereditary hemorrhagic telangiectasia (HHT) represents another vascular disorder closely linked to SMAD4 deficiency. The hallmark of HHT is loss or dysfunction of the capillary bed, leading to direct arteriovenous shunts (AVMs) between arteries and veins, and SMAD4 haploinsufficiency, caused by missense mutations and deletions of the Smad4 gene, is an established pathogenic mechanism underlying human HHT[19]. When AVMs occur in the lung, pulmonary AVMs generate shunts between pulmonary arteries and veins, thereby producing hemodynamic disturbances[31,32]. Previous studies[33,34] suggest that AVM formation predominantly involves venous/capillary ECs, with dysregulated endothelial polarization/migration and increased endothelial proliferation occurring preferentially within the venous-capillary vascular plexus, whereas arterial ECs are relatively less affected. In line with these observations, we show minimal changes in arterial ECs after Smad4 loss, but substantial transcriptomic shifts in venous/gCap EC subpopulations. Thus, these subpopulations may constitute the major EC subsets vulnerable to Smad4 loss-associated vascular malformations, thereby predisposing to the pathogenesis of HHT in the lung as well as in other organs.
aCap ECs appear to have a greater reliance on SMAD4 signaling, as they experience a more extensive remodeling phenotype in response to Smad4 loss. Under physiological conditions, aCap ECs remain largely quiescent and display a large, flattened morphology with extensive projections that intimately interdigitate with type I alveolar epithelial cells to form the gas diffusion surface[15,27]. Previous studies of the air-blood exchange interface have focused primarily on the alveolar epithelium, which is considered highly sensitive to proteostatic stress, energetic constraints, and mechanical disturbance[35-38]. By contrast, the contribution of aCap ECs to air-blood exchange interface stability has remained underexplored. Under baseline conditions, high expression of Car4 could support efficient CO2 conversion and gas exchange mediated by aCap ECs[15,16,39], whereas vascular endothelial growth factor 2 (VEGFR2) renders aCap ECs more responsive to VEGFA signals derived from type I alveolar epithelial cells, thereby maintaining aCap EC survival and normal alveolar architecture[40]. Upon Smad4 loss, however, aCap ECs showed upregulation of Car4, suggesting a state of functional compensation. However, under the continuous pressure of SMAD4 deficiency, this compensatory mechanism may become overloaded and shift to a maladaptive state, eventually leading to impaired aCap EC functions (e.g., oxygenation) in the lung. This may represent another potential mechanism underlying PH, anemia and/or heart failure phenotypes in Smad4-null mice[9,41]. In addition, genetic evidence has shown that SMAD4 deficiency impairs endothelial responsiveness to flow/BMP signaling and, by relieving BMP/ALK1-mediated inhibition of the PI3K/AKT pathway, may indirectly amplify downstream signaling output driven by VEGFR2[34]. Consistent with this model, our study revealed a marked upregulation of VEGFR2 expression in aCap ECs upon Smad4 loss, providing transcriptomic evidence for this effect. Smad4-null aCap cells also upregulated Ca2+ signaling and organic acid-binding pathways, accompanied by increased expression of endothelial junction-related gene sets. Given that CO2 diffusion is tightly coupled to local pH, HCO3- and organic acid gradients at the air-blood exchange interface, and that Ca2+ signaling is known to regulate endothelial permeability and barrier function[42,43], these changes strongly suggest that SMAD4 signaling critically contributes to acid-base and ionic microenvironment homeostasis at the alveolar-capillary surface. With respect to the clinical relevance of aCap EC-associated alterations, we note that pulmonary fibrosis is characterized by lung parenchymal injury and destabilization of the air-blood barrier. Although SMAD4 has not been reported as a frequently mutated gene in patients with idiopathic pulmonary fibrosis (IPF), genetic studies in animal models indicate that SMAD4 mutations, typically involving codons 496 or 500, cause Myhre syndrome, a progressive multisystem connective tissue disorder that can lead to pulmonary fibrosis[44]. Among the hub genes identified in our analysis, Hsp90, as well as other Hsp family members, has been implicated in IPF pathogenesis, as the HSP network can drive fibrotic progression by regulating proteostasis and cellular stress responses, thereby emerging as a potential therapeutic target[45]. Whether SMAD4 deficiency in aCap ECs plays a pathogenic role in certain forms of IPF warrants further investigation.
aCap and gCap ECs are interdigitated in a mosaic-like pattern within the alveolar gas-exchange region. They share a common developmental origin, but aCap ECs undergo postnatal specialization to acquire the highly efficient gas-exchange function at the air-blood exchange interface[15]. During vascular repair after acute lung injury, gCap ECs exhibit stem cell/progenitor features and serve as a source for capillary renewal, whereas aCap ECs are more terminally differentiated and their maintenance largely relies on gCap EC-derived replacement[15]. Given that our conclusions are drawn from a single-time-point scRNA-seq, the temporal relationship between the “vulnerable state” of venous/gCap ECs and the “immune-activated state” of aCap ECs remains unresolved. We propose that the immune-activated signature in aCap ECs may arise from both cell-intrinsic responses to Smad4 loss and extrinsic influences linked to venous/gCap EC remodeling, as well as epithelial niche perturbation. Dissecting the temporal ordering of these states will require longitudinal sampling and/or spatial transcriptomics.
In contrast to single-gene differential testing, hdWGCNA[12] captures coordinately regulated gene modules that shift in defined cellular states or disease contexts, thereby prioritizing candidate regulators at the systems level. Because direct network construction from single cells can be dominated by technical noise, hdWGCNA aggregates transcriptionally similar cells into metacells without disrupting subpopulation integrity, allowing extraction of shared features while preserving biological variation within cell types/subtypes for robust and biologically meaningful network inference. Among the 22 hub genes identified in the venous/gCap and aCap EC subpopulations, more than 50% may act as SMAD4 transcriptional targets or be directly regulated by SMAD4, including Atf3, Tbx3, Car4, Ccnl1, Egr1, Fos, Fosb, Hsp90aa1, Hspa2a, Ier3, Ubc, Rpl38, and Rps29. Among these candidates, Atf3 has previously been shown to participate in TGF-β-dependent transcriptional regulation and to be functionally coupled with SMAD4[46,47], whereas Tbx3, Egr1, and Fosb have been identified as direct transcriptional targets of SMAD4 in prior studies[48-50], supporting the ChIP-seq evidence observed here. The transcriptional control of Rpl38[51], Rps29[52], Ubc[53], and Hsp family members[54,55] may represent a critical mechanism connecting SMAD4 signaling to proteostasis regulation. Moreover, SMAD4 may indirectly regulate Vegfr2 expression via early growth response protein 1 (EGR1) and other co-regulators[56] and further influence the expression of the Vegfr2 effector Ptptrb (encoding vascular endothelial protein tyrosine phosphatase, VE-PTP)[57], or control Ppp1r15a expression (encoding growth arrest and DNA damage-inducible protein 34, GADD34) via ATF3[58]. SMAD4 can also interact with SMAD3, c-Jun and c-Fos to form an activator protein-1 (AP-1) transcription factor complex, thereby mediating TGF-β-induced transcription[59]. Therefore, SMAD4 deficiency may alter the composition and activity of the AP-1 complex, leading to transcriptional changes in downstream AP-1 target genes, such as Junb[59] and Ednrb[60,61]. Notably, although Car4 has not previously been reported as a SMAD4 target gene, our results suggest SMAD4-dependent transcriptional regulation on this important gene, raising a possibility that SMAD4 governs aCap cell fate and function through direct control of aCap identity-related genes.
Limitations
Several limitations of this study should be acknowledged. First, this work is based on a systematic re-analysis of a previously published mouse single-cell transcriptomic dataset without complementary protein-level measurements or functional validation; therefore, our conclusions primarily reflect transcriptome-level changes, and the causal relationships underlying gene regulation remain to be established in future studies. Second, because a whole-body inducible Smad4-null model was used, transcriptomic changes observed in SMAD4-deficient vascular ECs may be influenced by microenvironmental remodeling and aberrant intercellular communication from non-endothelial components such as smooth muscle cells or immune
Conclusion
In summary, our analyses delineate the transcriptomic responses of pulmonary vascular EC subpopulations to SMAD4 deficiency at single-cell resolution, which may mirror key pathophysiological features observed in multiple pulmonary vascular-related disorders, such as HHT and IPF. Therefore, we propose a vascular EC subpopulation-based model of SMAD4 regulation that may help explain the diverse responses of vascular segments under disease conditions and provide conceptual grounds for developing more specific therapeutic strategies for pulmonary vascular diseases.
DECLARATIONS
Author contributions
Study design: Zhou W, Chen Y, Zhang F
Study conduction: Zhou W, Zeng L
Manuscript writing: Zhou W, Zeng L, Chen Y, Zhang F
Availability of data and materials
All data are included in the manuscript and the Supplementary Materials. Additional information can be obtained from the corresponding authors upon reasonable request.
AI and AI-assisted tools statement
Not applicable.
Financial sponsorship
This project was supported by grants from NSFC (82425008, 82241009, 82070500) and National Key R&D Program of China (2021YFA1101200) to Feng Zhang, grants from NSFC (U23A20397, 82271609, 81770229 and 81970200) and Science and Technology Program of Guangzhou City (2023B01J1011, 2023A03J0724, 201707010206) to Yangxin Chen. Weibin Zhou was supported by grants from NSFC (82300559) and China Postdoctoral Science Foundation (2023TQ0396, 2024M753788).
Conflicts of interest
Zhang F is an Editorial Board Member of Vessel Plus, but was not involved in any steps of the editorial process, notably including reviewer selection, manuscript handling, or decision-making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Huertas A, Guignabert C, Barberà JA, et al. Pulmonary vascular endothelium: the orchestra conductor in respiratory diseases: highlights from basic research to therapy. Eur Respir J. 2018;51:1700745.
2. Travaglini KJ, Nabhan AN, Penland L, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature. 2020;587:619-25.
4. Kulikauskas MR, X S, Bautch VL. The versatility and paradox of BMP signaling in endothelial cell behaviors and blood vessel function. Cell Mol Life Sci. 2022;79:77.
5. Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92:645-56.
6. Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci USA. 1998;95:3667-72.
7. Lan Y, Liu B, Yao H, et al. Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol Cell Biol. 2007;27:7683-92.
8. Sirard C, de la Pompa JL, Elia A, et al. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev. 1998;12:107-19.
9. Lv W, Gu X, Zeng L, et al. Endothelial SMAD4 deficiency promotes pulmonary hypertension by impairing cell adhesion and extracellular matrix organization. Hypertension. 2025;82:1175-91.
10. Banerjee K, Lin Y, Gahn J, et al. SMAD4 maintains the fluid shear stress set point to protect against arterial-venous malformations. J Clin Invest. 2023;133:e168352.
11. Li F, Lan Y, Wang Y, et al. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev Cell. 2011;20:291-302.
12. Morabito S, Reese F, Rahimzadeh N, Miyoshi E, Swarup V. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell Rep Methods. 2023;3:100498.
13. Liu J, Cho SN, Akkanti B, et al. ErbB2 pathway activation upon Smad4 loss promotes lung tumor growth and metastasis. Cell Rep. 2015;10:1599-613.
14. dela Paz NG, D'Amore PA. Arterial versus venous endothelial cells. Cell Tissue Res. 2009;335:5-16.
15. Gillich A, Zhang F, Farmer CG, et al. Capillary cell-type specialization in the alveolus. Nature. 2020;586:785-9.
16. Schupp JC, Adams TS, Cosme C Jr, et al. Integrated single-cell atlas of endothelial cells of the human lung. Circulation. 2021;144:286-302.
17. Bai C, Fukuda N, Song Y, Ma T, Matthay MA, Verkman AS. Lung fluid transport in aquaporin-1 and aquaporin-4 knockout mice. J Clin Invest. 1999;103:555-61.
18. Sharma A, Niethamer TK. Specialized pulmonary vascular cells in development and disease. Annu Rev Physiol. 2025;87:229-55.
19. Al Tabosh T, Al Tarrass M, Tourvieilhe L, Guilhem A, Dupuis-Girod S, Bailly S. Hereditary hemorrhagic telangiectasia: from signaling insights to therapeutic advances. J Clin Invest. 2024;134:e176379.
20. Crist AM, Lee AR, Patel NR, Westhoff DE, Meadows SM. Vascular deficiency of Smad4 causes arteriovenous malformations: a mouse model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis. 2018;21:363-80.
21. Mora Massad K, Dai Z, Petrache I, Ventetuolo CE, Lahm T. Lung endothelial cell heterogeneity in health and pulmonary vascular disease. Am J Physiol Lung Cell Mol Physiol. 2025;328:L877-84.
22. Wu M, Wu S, Chen W, Li YP. The roles and regulatory mechanisms of TGF-β and BMP signaling in bone and cartilage development, homeostasis and disease. Cell Res. 2024;34:101-23.
23. Hiepen C, Mendez PL, Knaus P. It takes two to tango: endothelial TGFβ/BMP signaling crosstalk with mechanobiology. Cells. 2020;9:1965.
24. Goumans MJ, Valdimarsdottir G, Itoh S, et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817-28.
25. Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6:16-26.
26. Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest. 2016;126:821-8.
27. Trimm E, Red-Horse K. Vascular endothelial cell development and diversity. Nat Rev Cardiol. 2023;20:197-210.
28. Icli B, Feinberg MW. Plasticity of arterial and venous endothelial cell identity: some nerve! Circ Res. 2016;119:574-6.
29. Ma L, Chung WK. The role of genetics in pulmonary arterial hypertension. J Pathol. 2017;241:273-80.
30. Richter A, Yeager ME, Zaiman A, Cool CD, Voelkel NF, Tuder RM. Impaired transforming growth factor-beta signaling in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;170:1340-8.
31. Tellapuri S, Park HS, Kalva SP. Pulmonary arteriovenous malformations. Int J Cardiovasc Imaging. 2019;35:1421-8.
32. Gossage JR, Kanj G. Pulmonary arteriovenous malformations. A state of the art review. Am J Respir Crit Care Med. 1998;158:643-61.
33. Park H, Furtado J, Poulet M, et al. Defective flow-migration coupling causes arteriovenous malformations in hereditary hemorrhagic telangiectasia. Circulation. 2021;144:805-22.
34. Ola R, Künzel SH, Zhang F, et al. SMAD4 prevents flow induced arteriovenous malformations by inhibiting casein kinase 2. Circulation. 2018;138:2379-94.
35. Romero F, Summer R. Protein folding and the challenges of maintaining endoplasmic reticulum proteostasis in idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2017;14:S410-3.
36. Sanches Santos Rizzo Zuttion M, Moore SKL, Chen P, Beppu AK, Hook JL. New insights into the alveolar epithelium as a driver of acute respiratory distress syndrome. Biomolecules. 2022;12:1273.
37. Katzen J, Beers MF. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J Clin Invest. 2020;130:5088-99.
38. Leiby KL, Raredon MSB, Niklason LE. Bioengineering the Blood-gas Barrier. Compr Physiol. 2020;10:415-52.
39. Sun X, Perl AK, Li R, et al. A census of the lung: CellCards from LungMAP. Dev Cell. 2022;57:112-45.e2.
40. Vila Ellis L, Cain MP, Hutchison V, et al. Epithelial vegfa specifies a distinct endothelial population in the mouse lung. Dev Cell. 2020;52:617-30.e6.
41. Machiya A, Tsukamoto S, Ohte S, Kuratani M, Suda N, Katagiri T. Smad4-dependent transforming growth factor-β family signaling regulates the differentiation of dental epithelial cells in adult mouse incisors. Bone. 2020;137:115456.
42. Townsley MI. Permeability and calcium signaling in lung endothelium: unpack the box. Pulm Circ. 2018;8:2045893217738218.
43. Ying X, Minamiya Y, Fu C, Bhattacharya J. Ca2+ waves in lung capillary endothelium. Circ Res. 1996;79:898-908.
44. Lin AE, Scimone ER, Thom RP, et al. Emergence of the natural history of Myhre syndrome: 47 patients evaluated in the Massachusetts General Hospital Myhre Syndrome Clinic (2016-2023). Am J Med Genet A. 2024;194:e63638.
45. Roque W. Heat shock proteins in pulmonary fibrosis: pawns of cell homeostasis. Am J Physiol Cell Physiol. 2022;322:C1105-9.
46. Rohini M, Arumugam B, Vairamani M, Selvamurugan N. Stimulation of ATF3 interaction with Smad4 via TGF-β1 for matrix metalloproteinase 13 gene activation in human breast cancer cells. Int J Biol Macromol. 2019;134:954-61.
47. Kang Y, Chen CR, Massagué J. A self-enabling TGFbeta response coupled to stress signaling: smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003;11:915-26.
48. Gamart J, Barozzi I, Laurent F, et al. SMAD4 target genes are part of a transcriptional network that integrates the response to BMP and SHH signaling during early limb bud patterning. Development. 2021;148:dev200182.
49. Fei T, Xia K, Li Z, et al. Genome-wide mapping of SMAD target genes reveals the role of BMP signaling in embryonic stem cell fate determination. Genome Res. 2010;20:36-44.
50. Li J, Weinberg MS, Zerbini L, Prince S. The oncogenic TBX3 is a downstream target and mediator of the TGF-β1 signaling pathway. Mol Biol Cell. 2013;24:3569-76.
51. Gopanenko AV, Kolobova AV, Meschaninova MI, et al. Knockdown of the mRNA encoding the ribosomal protein eL38 in mammalian cells causes a substantial reorganization of genomic transcription. Biochimie. 2021;184:132-42.
52. Xu W, Guo Z, Guan Y, et al. Machine learning-based proteomics profiling of ALS identifies downregulation of RPS29 that maintains protein homeostasis and STMN2 level. Commun Biol. 2025;8:1177.
53. Yang X, Lan T, Zhang B, et al. Targeting ubiquitination in disease and therapy. Signal Transduct Target Ther. 2025;10:424.
54. Chiosis G, Digwal CS, Trepel JB, Neckers L. Structural and functional complexity of HSP90 in cellular homeostasis and disease. Nat Rev Mol Cell Biol. 2023;24:797-815.
55. Kotler JLM, Street TO. Mechanisms of protein quality control in the endoplasmic reticulum by a coordinated Hsp40-Hsp70-Hsp90 system. Annu Rev Biophys. 2023;52:509-24.
56. Jung E, Ou S, Ahn SS, Yeo H, Lee YH, Shin SY. The JNK-EGR1 signaling axis promotes TNF-α-induced endothelial differentiation of human mesenchymal stem cells via VEGFR2 expression. Cell Death Differ. 2023;30:356-68.
57. Baccouche B, Lietuvninkas L, Kazlauskas A. Activin A limits VEGF-induced permeability via VE-PTP. Int J Mol Sci. 2023;24:8698.
58. Jiang HY, Wek SA, McGrath BC, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. 2004;24:1365-77.
59. Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature. 1998;394:909-13.
60. Cattaruzza M, Eberhardt I, Hecker M. Mechanosensitive transcription factors involved in endothelin B receptor expression. J Biol Chem. 2001;276:36999-7003.
61. Wagner AH, Krzesz R, Gao D, Schroeder C, Cattaruzza M, Hecker M. Decoy oligodeoxynucleotide characterization of transcription factors controlling endothelin-B receptor expression in vascular smooth muscle cells. Mol Pharmacol. 2000;58:1333-40.
62. Zhang P, Hou S, Chen J, et al. Smad4 deficiency in smooth muscle cells initiates the formation of aortic aneurysm. Circ Res. 2016;118:388-99.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].