

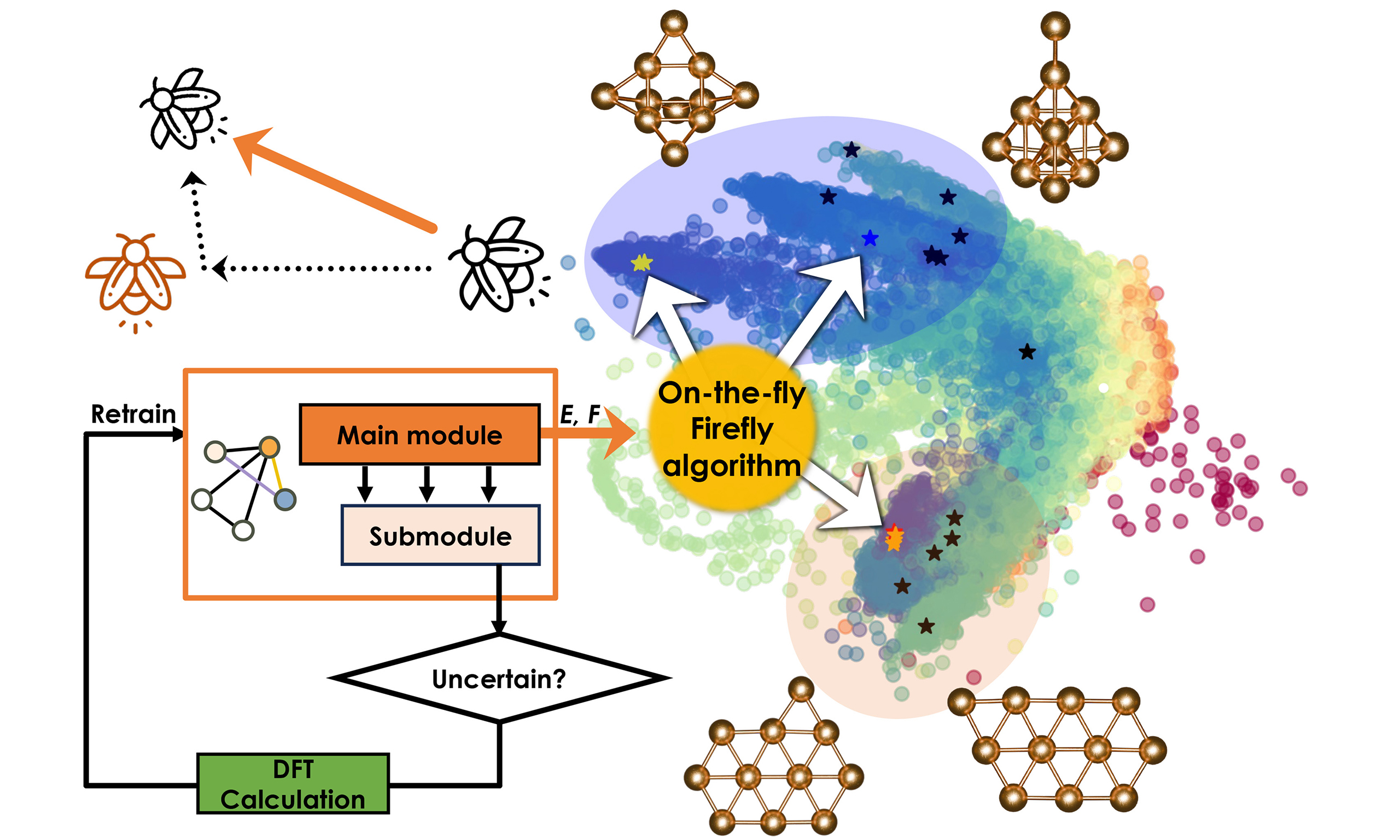
Robust global optimization of atomic structures via a learning loss-informed on-the-fly firefly algorithm
 0
0 
Abstract
In computational materials science, global optimization is pivotal for bridging theory and experiment but can fail when the theoretical treatment defining the potential energy surface does not accurately predict stability trends. Conventional approaches to address this rely on statistical sampling over numerous independent calculations or the use of more expensive theories throughout the global optimization process, both of which substantially increase computational cost. To overcome this, we present nature-inspired algorithm for robust atomic structure search (NARA), a framework that combines a firefly algorithm-based multimodal search with uncertainty-aware active learning. Instead of converging to a single structure, NARA simultaneously explores multiple distinct configurations, thereby mitigating sensitivity to potential limitations. For the “8” surface oxide on Cu(111), it achieves higher efficiency than the widely used basin-hopping algorithm. For gold clusters, a single run recovers both planar and non-planar structures, resolving stability reversals induced by different theoretical treatments. NARA thus achieves both efficiency and robustness for reliable atomic-structure identification.
Keywords
INTRODUCTION
Identifying the accurate atomic structure of materials is critically important for materials science and catalysis because the stability, reactivity, and functionality of materials strongly depend on their atomic configurations[1-4]. For example, when certain atomic structures show enhanced performance in the CO2 reduction reaction (CO2RR), failure to understand the key structural factors behind this improvement can lead to non-reproducible results, making the findings merely incidental observations. Conversely, clear structural insights, such as the better CO2RR performance of Cu2O(110) facets compared to Cu2O(100) or Cu2O(111) facets[5-7], or the beneficial effect of subsurface oxygen on CO2RR[8-10], provide scientific milestones and practical design rules for subsequent studies.
The recognition of the importance of determining accurate atomic structures has driven significant advancements in experimental characterization techniques. Cutting-edge surface science tools such as scanning tunneling microscopy (STM) and non-contact atomic force microscopy (nc-AFM) now provide atomic-scale resolution, allowing detailed visualization of catalyst surfaces and reaction intermediates[11]. These techniques have profoundly improved our ability to relate atomic-scale structures to improve catalytic properties.
Despite these remarkable advances, significant limitations remain. First, experimental characterization methods are typically carried out under conditions far from actual catalytic operating environments. Due to the inherent difference between experimental and real reaction conditions, observed atomic structures may differ substantially from those relevant to catalytic performance[12]. Second, unwanted adsorbates, impurities, or reaction by-products can obscure or alter surface structures, making direct observations challenging or even misleading. Third, experimental characterization is fundamentally only available under the physical existence of the materials, thereby limiting its applicability to those structures that already exist. Hypothetical materials thus remain inaccessible by experimental characterization alone.
Given these limitations, theoretical methods have emerged as essential tools to complement experimental approaches. In the beginning, researchers modeled structures based on prior knowledge or physical and chemical intuition[2]. Subsequently, due to the increasing necessity of considering a high degree of freedom, more systematic global optimization algorithms such as basin hopping, genetic algorithms, and particle swarm optimization methods have been introduced to explore the potential energy surface (PES) effectively without prior structural assumptions[13-15]. Furthermore, incorporating machine learning techniques has dramatically improved the efficiency of these global optimization algorithms by significantly reducing the number of expensive target function [mostly density-functional theory (DFT)] calculations[16-25].
However, machine learning-based energy predictions inherently involve uncertainties. If global optimization is performed using machine learning models trained on incomplete or inadequately sampled PESs, it may fail to approach the true global minimum or lead to computational instabilities, ultimately causing optimization failures. To address these challenges, state-of-the-art global optimization frameworks often incorporate active-learning schemes, systematically evaluating the uncertainty of generated structures to enhance robustness and reliability[20,22-24].
Nevertheless, even robust active learning methods cannot entirely mitigate uncertainties originating from the chosen target potential itself. Specifically, if the selected target potential inadequately captures energy trends among competing metastable states, the optimization results may not reflect the true global minimum on the PES. Such cases frequently occur when additional theoretical treatments, such as temperature effects, surrounding environments, van der Waals (vdW) corrections, or magnetism, must be considered to accurately represent the system[26,27]. Under these conditions, no matter how robust the optimizer or accurate the machine learning model is, active learning alone cannot fully address this fundamental limitation.
To resolve such issues, two general approaches can be taken. The former is to perform global optimization calculations that explicitly incorporate the additional theoretical treatments. While this can improve accuracy, it also results in a significantly increased computational cost, often to the point of being impractical. The latter is to conduct multiple independent calculations to statistically analyze metastable competing states. However, this strategy can easily overlook relevant metastable structures, as most global optimization algorithms are narrowly directed toward locating only the global minimum.
A clear and intuitive example of this challenge is provided by the study of gold clusters reported in Ref.[26]. The authors demonstrated that the inclusion of vdW or many-body dispersion (MBD) corrections could alter the predicted stable structure of Au11 and Au13 clusters, favoring three-dimensional (3D) non-planar configurations over two-dimensional (2D) planar shapes predicted by standard DFT calculations using the generalized gradient approximation (GGA) alone.
To overcome these limitations, we propose nature-inspired algorithm for robust atomic structure search (NARA), a global optimization framework based on the FA. Unlike conventional approaches that typically report a single global minimum, NARA identifies multiple structurally distinct local minima in addition to the global minimum. This approach provides a robust optimization strategy that is inherently less sensitive to the choice of theoretical treatments, thereby offering an efficient and reliable method for identifying stable atomic structures under realistic and diverse conditions.
EXPERIMENTAL
DFT
All DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP)[28,29] with the projector augmented wave (PAW) method[30] and the GGA proposed by Perdew, Burke, and Ernzerhof (PBE)[31] as the exchange-correlation (xc) functional. A planewave kinetic energy cutoff of 500 eV was used. The convergence criteria for the electronic step were set to 10-5 eV, while structural relaxations were considered converged once the norm of forces on each atom was reduced to less than 10-2 eV/Å.
For the O/Cu(111) surface oxide calculations d, the DFT data was adopted from the database developed in our previous work[16]. All Cu(111) slab models in the database consist of four atomic layers, where the bottom two layers were fixed to represent a bulk-like region, and a minimum vacuum spacing of 14 Å was used along the surface-normal direction. A Γ-centered k-point grid with a spacing of 0.15 Å-1 was used consistently throughout all the structures within the database. For the gold cluster calculations, a cubic unit cell with dimensions of 20 Å along each axis was used, and all atoms are set apart from the unit cell boundary more than 5 Å to avoid the unwanted interactions between repeated unit cells. The 5d10 6s1 orbitals of gold atoms are considered as valence states, and all calculations were performed at the Γ-point. Dispersion corrections (e.g., vdW or MBD) were not applied, consistent with the aim of this work to assess robustness against target-potential limitations. Additional computational details (i.e., input settings) are provided in the Supplementary Materials.
Random structure generation and structure conditioning
For each structure, atomic coordinates (x, y, z) were randomly sampled from a uniform probability distribution within predefined regions or unit cells according to the required number of atoms. Because both random initialization and subsequent firefly algorithm (FA) update steps can generate candidate structures with unphysical close contacts, we apply a conditioning step to remove severe overlaps before energy/force evaluation. Such unrealistically short interatomic distances can destabilize calculations and increase computational costs, and this issue becomes more pronounced when using machine learning-based energy/force predictions due to the lack of training data in such regions.
To mitigate this, we employed a conditioning procedure applied immediately after random structure generation and after each FA update. In this procedure, atoms closer than a predefined threshold distance were repelled from each other before energy/force evaluation and subsequent local relaxation. We define this threshold using the sum of covalent radii[32] as a physically motivated length scale, and we set it to 80% of the summed radii as a practical compromise. This choice removes clearly unphysical close contacts while avoiding an overly conservative constraint that would unnecessarily inflate initial separations and bias the search. We emphasize that this conditioning is not enforced during the subsequent local relaxation under the target potential and therefore does not impose a hard lower bound on final bond lengths. We utilized Hooke’s law to define the repulsive force and corresponding energy whenever two atoms approach within the threshold[33] as follows,
Here, interatomic distances, rij, were computed using the neighbor analysis function implemented in the atomic simulation environment (ASE[34]), and structural relaxation was conducted to minimize the defined repulsive force. For this study, conditioning thresholds were set as follows: 2.18 Å for gold cluster and 1.58, 2.11, and 1.06 Å for Cu–O, Cu–Cu, and O–O, respectively, for O/Cu(111) surface oxides. As the defined conditioning forces and energies were not simultaneously combined with actual potential energies or atomic forces, a consistent spring constant of k was applied as 1 eV/ Å2 throughout this study.
Visualization of PES
To analyze the results of global optimization, the PES was post-processed and visualized. All structures generated during the global optimization were collected and used to train a variational autoencoder (VAE)-based architecture[35,36], reducing dimensionality to a 2D latent space for visualization. In defining global descriptors from symmetry-invariant local descriptors assigned to each atom, an attention pooling layer[37] was implemented. This layer computes attention scores from each atom’s local symmetry invariant descriptor and applies a softmax function to aggregate these into global descriptors. The training was conducted in two steps: initially, the attention pooling layer and VAE architecture were trained simultaneously to minimize the reconstruction error; subsequently, only the VAE parameters were optimized based on the evidence lower bound (ELBO)[35]. This approach ensured that attention scores highlighted structurally significant distinctions, and resulted in a continuous representation within the VAE latent space as well.
Implementation of the FA for atomic structure optimization
The FA operates according to three key principles: (i) all fireflies are treated as unisex, meaning any individual can be attracted to any other regardless of “gender”; (ii) mutual attractiveness is proportional to perceived brightness, which decays exponentially with distance; and (iii) each firefly moves toward the one it perceives as most attractive, or performs a random walk if no firefly is identified as attractive. When applied to atomic structure optimization, each firefly represents an atomic configuration, and the evolution of configurations toward the next generation is governed by[38]:
where xit denotes the Cartesian coordinates of agent i at generation t (a 3N representation for N atoms), xjt is the configuration of a more attractive agent j, and ε is a random displacement. The parameter α sets the step size of the random displacement term for agent i, and in atomic structure optimization it can be chosen analogously to the perturbation magnitude used in basin hopping. In this work, we use α =0.5 Å. The parameter β0 is the attractiveness at zero distance and sets the maximum scale of the attractiveness term. It is commonly treated as a fixed constant close to unity[38], and we use β0 =0.9. The parameter γ controls the distance-dependent decay of attractiveness, β(rij) = β0exp(-γrij2), and thus sets the effective interaction range between agents. A larger γ strengthens the distance decay and limits interactions to nearby agents, whereas a smaller γ maintains longer-ranged interactions. Accordingly, we keep α and β0 fixed across systems and adjust γ when the distance scale or energy-landscape characteristics change. Following prior FA studies that report practical population size in the range of 15-50[38], we use 30 agents to balance exploration diversity against the cost of local relaxations and energy/force evaluations.
In practice, the displacement term (xjt - xit) in Equation (2) cannot be taken as a direct subtraction of the raw 3N coordinate vectors, because two configurations that represent the same motif may differ by atom-index permutations and symmetry-related transformations. To obtain a meaningful displacement, we define (xjt - xit) only after establishing a consistent atom-to-atom correspondence between the two structures using our symmetry-invariant local descriptor-based atom index matching.
To effectively implement the FA with a proper association of atom indices, our method begins by selecting the most similar atom pair (a1, b1) from two distinct configurations, Structures A and B, respectively. This selection is based on a symmetry-invariant descriptor, which is obtained by extracting the “0e” channel from the symmetry-equivariant node features after the message-passing blocks. The optimal pair is then identified by minimizing the difference between their descriptors:
where ϕaA, ϕbB are symmetry-invariant descriptors of a1 and b1, respectively.
Subsequently, we calculate neighbor lists
From these neighbor lists, we select two pairs (a2, b2) and (a3, b3) by identifying the combination that minimizes the sum of their descriptor distances:
To ensure physical consistency, we further applied geometric criteria by comparing lengths of triangles formed by atom triplets (a1, a2, a3) and (b1, b2, b3), with tolerance of 0.5 Å. If no triplet satisfying the triangle tolerance is found after testing candidate combinations, only translation is applied.
We then compute the rotation matrix, R, required to align vectors (
• For structures with periodic boundary conditions (PBCs), we first identify the closest available rotation matrix from the list of possible rotations calculated through spglib[39].
• If spglib provides only the identity matrix, we first convert the rotation matrix to fractional coordinates with the cell matrix, L, as given by:
then round this fractional rotation matrix, Rfrac, to the nearest integer values, and check its orthogonality for rigorous lattice transformation.
• For non-PBC systems, the computed rotation matrix is used directly.
Once the appropriate rotation is determined, we apply it to initially align atom b1 in Structure B with a1 in Structure A. This provides a starting point for a full permutation alignment using the Hungarian algorithm[40]. We then perform a translation of the entire structure to minimize atom-wise root-mean-square deviation (RMSD), with an additional rotation applied for non-PBC systems. Finally, we calculate the atom-wise displacement vectors required to move each atom in Structure A to its corresponding position in the aligned Structure B.
This symmetry-invariant descriptor-informed structural alignment provides a well-defined displacement for atomic structures within our FA-based NARA framework.
Integration of learning loss into graph neural network interatomic potential
As depicted in Figure 1, the main module comprises multiple (usually 3 to 5) message-passing blocks, followed by an energy prediction block, which yields per-atom energies from symmetry-invariant “0e” scalar channels of node features. For periodic systems, we construct the pairwise distance matrix by explicitly including across-boundary atom pairs to ensure consistency under PBCs, as detailed in Supplementary Figures 1 and 2. Atomic forces are computed by differentiating the predicted energies with respect to atomic positions. So, the main module’s loss,
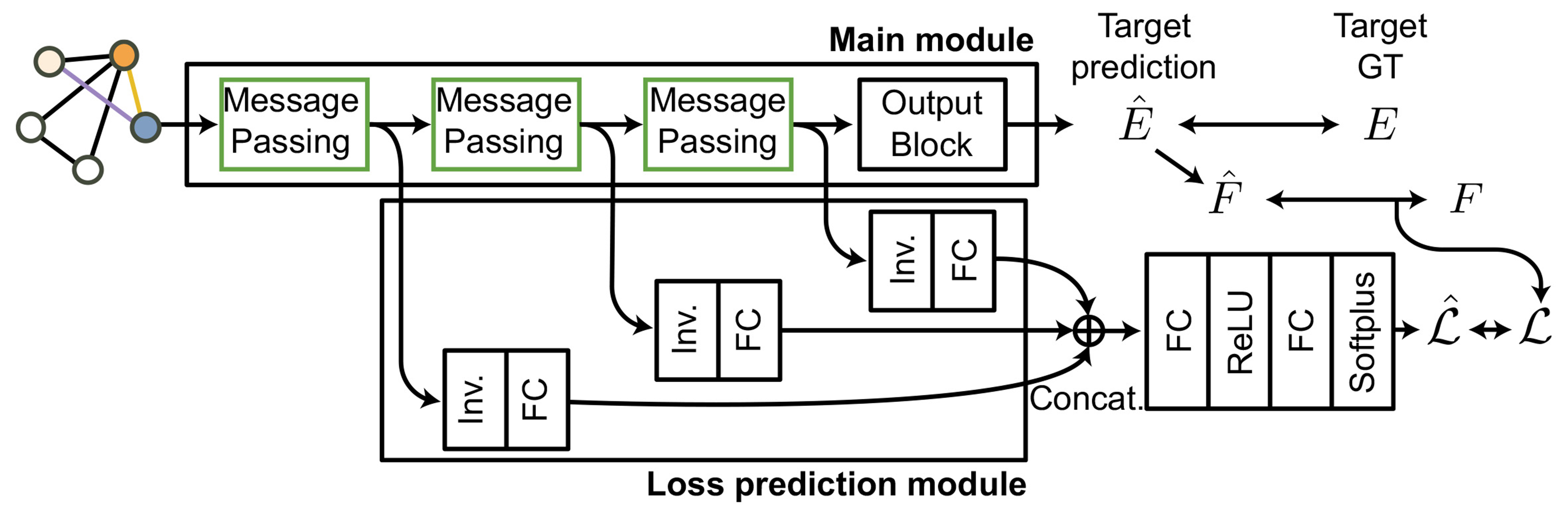
Figure 1. Architecture of GNN used in this work consists of a main module responsible for predicting energy and forces, and a loss prediction module that estimates the predicted loss,
where
To integrate learning loss into this main prediction module, we attached a loss-prediction submodule after minor modification. Specifically, instead of employing global average pooling layers, we extracted symmetry-invariant (“0e”) node features from each message-passing block and concatenated these features to predict the loss corresponding to the deviation of atomic forces between prediction and reference. The submodule was trained using the margin ranking loss approach described in the original paper[42], pairing two atomic environments within each batch to ensure consistency between the trends of predicted loss,
where y equals +1 if
The uncertainty-quantification performance of the learning loss approach, together with an assessment of whether introducing the learning loss submodule affects energy and force prediction, is reported in Supplementary Figures 3 and 4. The associated computational overhead is summarized in Supplementary Table 1.
RESULTS AND DISCUSSION
NARA framework for robust global optimization of atomic structures
FAs for multimodal optimization
To enhance robustness against variations in the theoretical treatments of the target potential, we adopted the FA[38], a population-based metaheuristic evolutionary algorithm known for its strong performance in multimodal optimization problems. A distinguishing characteristic of the FA compared to other global optimization algorithms is the exponential attenuation of brightness with distance, which prevents convergence to a single minimum when structures are significantly separated in configuration space. As a result, the FA naturally facilitates multimodal optimization by simultaneously identifying multiple structurally distinct local minima in addition to the global minimum.
Previous studies have shown that the FA not only efficiently locates multiple local minima but also demonstrates superior or comparable global optimization performance relative to other evolutionary algorithms[38]. For instance, prior applications of FA-based optimization successfully identified global minima in phosphorus structures[43] and Al42- clusters[44]. While these results highlight the accuracy and versatility of the FA, practical implementations still face two major limitations: (i) computational inefficiency due to the use of expensive potentials such as DFT or density-functional tight-binding (DFTB) throughout the entire optimization process; and (ii) difficulties in properly associating atoms, when generating or comparing structures within FA-based optimization under symmetry transformations such as rotation, reflection, inversion, and translation.
E(3)-equivariant GNN interatomic potential with learning loss
To address the first limitation, we adopted an E(3)-equivariant GNN based on the neural equivariant interatomic potentials (NequIP)[41], known for achieving high accuracy with relatively small training samples. To ensure the reliable and robust predictions of the main module, uncertainty quantification was adopted on top of the main prediction module. Although the ensemble-based query-by-committee (QBC) method is commonly used for uncertainty quantification in this field[45-47], and generally outperforms the single model-based approaches[48], it requires the simultaneous evaluations of multiple models, significantly increasing computational costs for both training and inference.
To effectively address this limitation, we adopted the learning loss method proposed by Yoo et al.[42]. The key idea of this method is to attach a compact submodule to the main module, which predicts the loss of the main module while the main module predicts the target values. This approach yields reliable, task-agnostic uncertainty estimates without significant architectural modifications. In our implementation, the main module predicts per-atom energies and forces for atomic structures, whereas the submodule estimates the difference between the predicted energy and the target energy computed by the reference potential (e.g., DFT). Unlike QBC methods, which rely on multiple model ensembles, our approach integrates only a compact submodule into a single GNN model, thereby significantly reducing computational costs compared to QBC-based methods. For these reasons, we adopted the learning loss approach with minor modifications to align the original architecture with our GNN model, as illustrated in Figure 1. All implementations are carried out in the in-house LLUMYS (Learning-Loss based Uncertainty-aware MLIP from Yonsei & Sydney) code (see Availability of data and materials), and further architectural details are provided in the Methods.
Evolution of atomic structures in the FA
The second limitation concerns the structural evolution step in the FA, which requires a consistent atom-to-atom mapping to define meaningful displacement vectors. In prior works[43,44], non-periodic structures were aligned by translating to a common center-of-mass, applying rigid-body rotation using the Kabsch algorithm[49], and performing permutation matching via the Hungarian algorithm[40]. However, under PBCs, these methods often neglect symmetry operations such as rotation, reflection, or inversion. As illustrated in Figure 2, when Structure B is symmetry-transformed from Structure A, the resulting displacement vectors become inaccurate. Although identical structures can be filtered using symmetry-invariant fingerprints, such global approaches fail for cases where structures are mostly similar but contain localized differences, leading to a physically inaccurate representation of atomic displacements and reducing the efficiency of structural evolution.
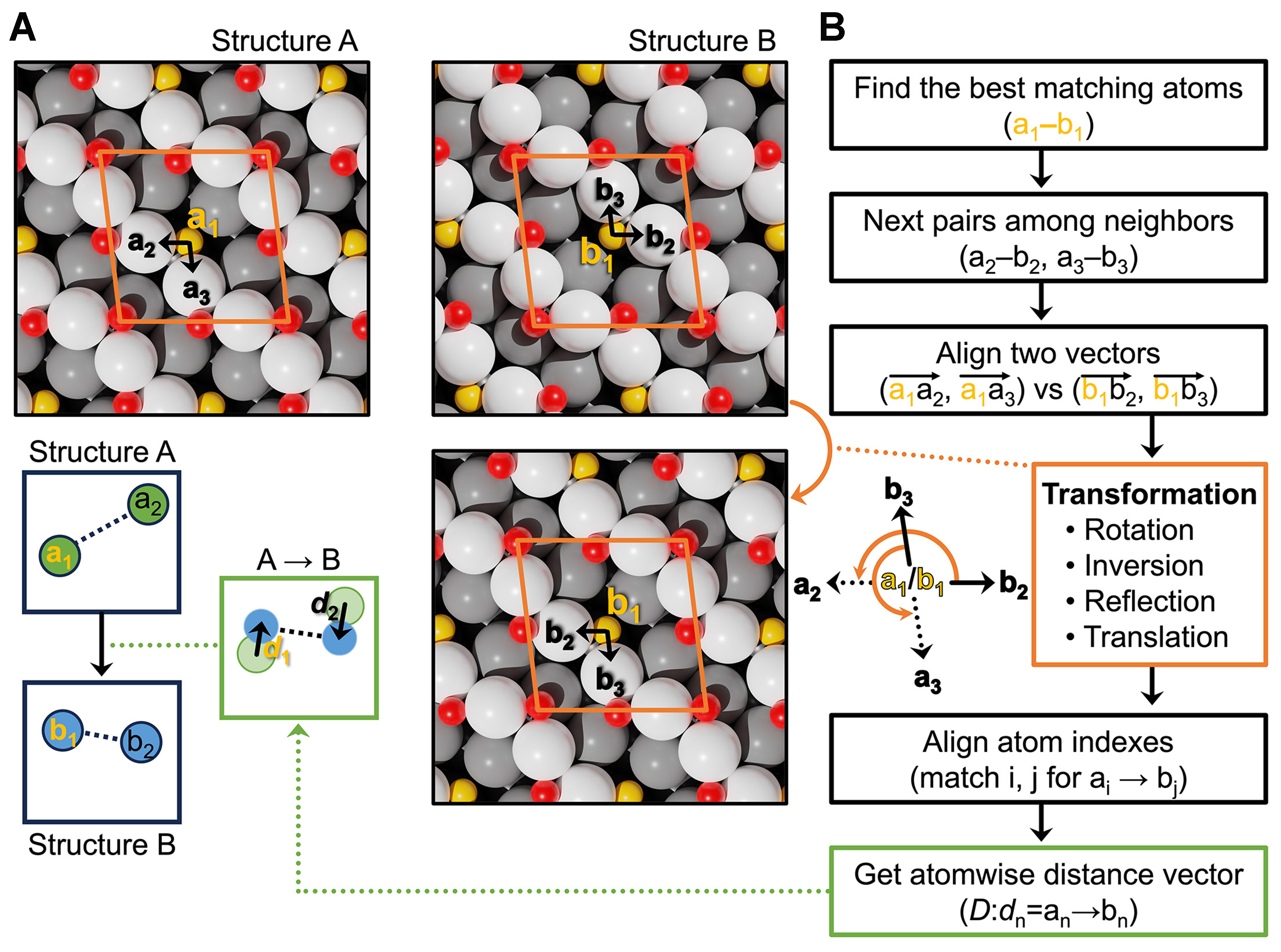
Figure 2. Figure 2. (A) Example of structural matching between Structure A and Structure B during the FA. Structure B is modified using symmetry operations to minimize the RMSD relative to Structure A; (B) Flowchart of structure evolution in the FA. FA: Firefly algorithm; RMSD: root-mean-square deviation.
To resolve this, we incorporated per-atom symmetry-invariant descriptors extracted from the GNN, enabling atom matching based on local environments rather than atomic coordinates. The descriptor-informed approach ensures that interpolations between structures remain physically meaningful and symmetry-consistent even under PBC. Figure 2 outlines the procedure, while detailed algorithms and mathematical formulations are provided in the Methods. All implementations are available in the NARA code (see Code availability).
On-the-fly FA searches
As previously discussed, we implemented an active learning framework as depicted in Figure 3, to enhance the robustness of our machine learning-accelerated global optimization. This framework automatically performs calculations using the target potential (e.g., DFT) based on the uncertainty quantifications provided by the learning loss submodule attached to the GNN main module, subsequently retraining the GNN to ensure reliability. Here, a critical aspect of this framework is deciding which structures to recalculate.
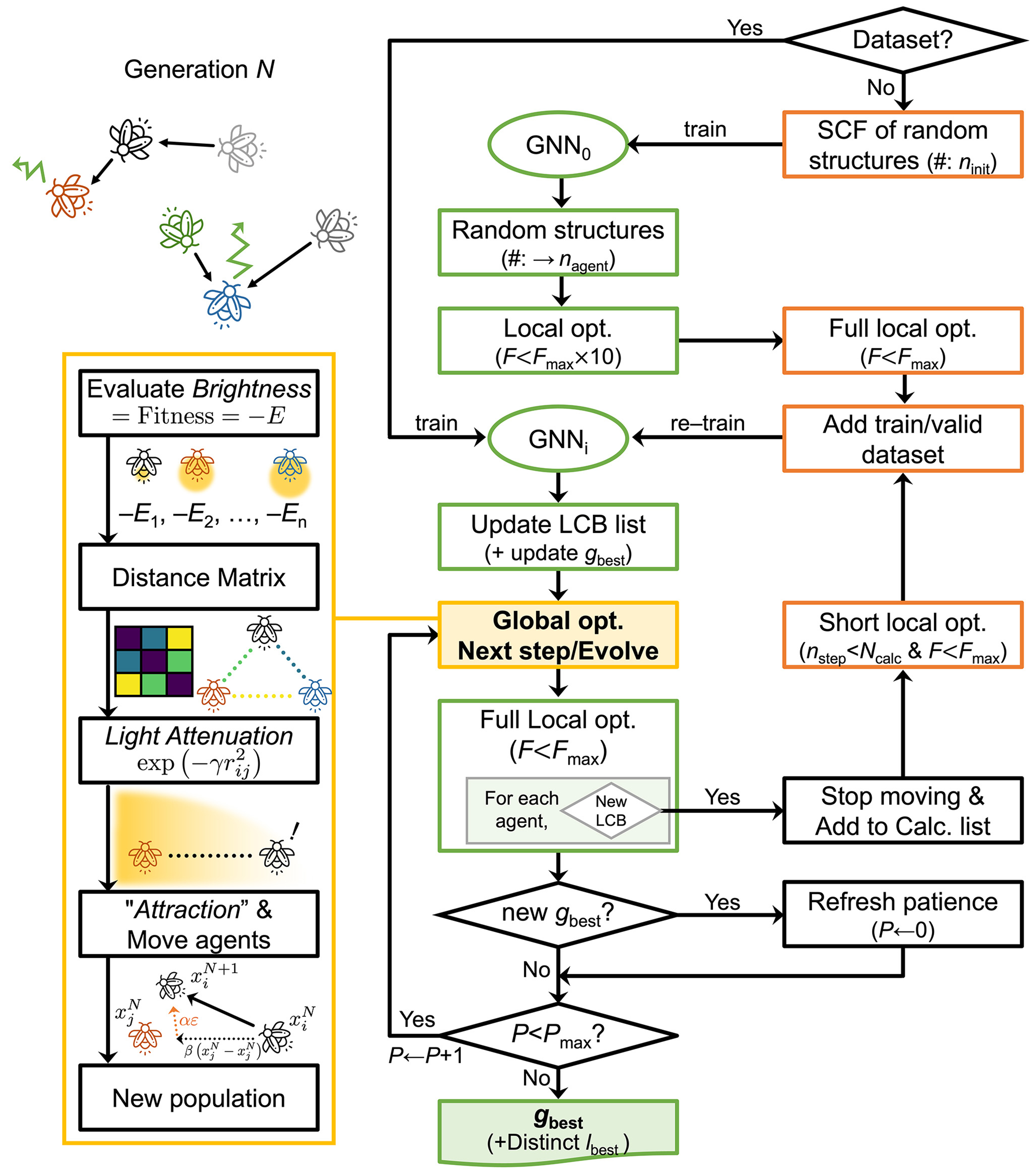
Figure 3. The active learning procedure implemented in NARA. The green-colored boxes in the middle column represent energy evaluations performed using the GNN, while the orange-colored boxes in the right column correspond to energy evaluations based on the target potential (e.g., DFT). NARA: Nature-inspired algorithm for robust atomic structure search; GNN: graph neural network; DFT: density-functional theory; SCF: self-consistent field calculations; LCB: lower-confidence-bound.
In molecular dynamics simulations accelerated by machine learning interatomic potentials (MLIPs), a common active learning strategy is recalculating structures whose uncertainty exceeds a predefined threshold[45-47,50]. Even though it is effective for improving calculation stability, this approach does not align well with the goal of global optimization, which focuses primarily on identifying lower-energy structures. Indeed, results from previous work[51] indicate a significant drop in success rates when retraining is driven predominantly by uncertainty alone. Therefore, we adopted a lower-confidence-bound (LCB) approach[51,52] to balance thermodynamic stability and structural uncertainty. Specifically, structures that update the lowest LCB values are selected for recalculation. The LCB for a given structure X is defined as follows,
Here,
where the index k denotes the element type, and jk represents the set of atoms corresponding to element type k. The coefficients ak and bk are the parameters obtained from linear fitting for each element.
With the defined LCB, our active learning procedure follows these steps: In the absence of an initial dataset, we start by generating ninit random structures to build an initial GNN model (GNN0). We then generate other random structures, relax them with loose convergence criteria (using a convergence criteria ten times looser than the standard force convergence criteria), and employ k-means clustering to select representative structures. These structures are fully optimized using the target potential to form the initial training dataset for GNN (GNN1). If an initial dataset already exists, it directly constructs the first GNN model (GNN1). Subsequently, the trained GNN guides structural evolution within the FA through iterative generations. Each generational evolution comprises five stages, as detailed in the orange box (lower left panel of Figure 3): (a) Assess brightness (-E) for all agents; (b) Calculate a distance matrix among agents based on the differences between global descriptors, obtained by summing the per-atom features; (c) Evaluate each agent’s most attractive partner by implementing brightness attenuation as a function of distance, ensuring minimal interaction between distant agents; (d) Agents move towards their respective most attractive neighboring agents, generating new structures according to the FA’s evolution rule; (e) After relaxing newly generated structures, their LCBs are evaluated. If new minima are identified, those agents are frozen and do not participate in communication with other agents. Once the number of frozen agents reaches a predefined threshold, recalculations using the target potential are performed. The resulting structures are then added to the training and validation datasets, followed by retraining of the GNN. This iterative process continues until the global best solution remains unchanged for a predefined patience limit, Pmax.
Through this comprehensive procedure, our framework enables robust, machine learning-based global optimization even without an initial dataset. We validated its effectiveness in the gold cluster systems which is discussed later in this work.
Application: the “8” surface oxide on Cu(111)
We evaluated the efficiency of our proposed FA-based NARA framework in identifying the global minimum structure of the previously found “8” surface oxide on Cu(111) reported in our earlier work[16]. Basin hopping (BH) was chosen as the comparison method because it is a well-established global optimization approach for atomistic structure searches, including recent applications to complex surface systems[11]. It also serves as a conceptually clear reference for the non-communicating limit of the FA when the attractiveness term becomes negligible at large γ. We therefore use BH as a representative reference method for evaluating the efficiency of our FA-based framework in this case study. The same GNN model trained earlier for uncertainty quantification was employed here. Since our previous study confirmed the capability to accurately find the global minimum using this dataset, active learning was turned off, and only the GNN model was used for energy/force evaluations without the attached submodule.
For the comparison, BH was configured with a maximum atom-wise displacement of 0.5 Å, employing a Metropolis acceptance criterion at a temperature of T = 300 K. The FA was set up with 30 firefly agents, using parameters α =0.5, β0 = 0.9, γ = 0.02. The trajectory of FA agents on the PES is visualized in Figure 4A. The PES was constructed by training a VAE-based neural network with local attention scores, using all structures encountered during the FA search, mapping them into a 2D latent space. Energy levels are indicated by colors, and the known global minimum, “8”@2023t is marked with a white star as a reference. Initially, at the first generation (Gen. 1), the agents, marked with black stars, are located in higher-energy regions since they are just local optimized structures after random initialization. From the second generation (Gen. 2) onward, the evolutionary trajectories which are depicted by black arrows, illustrate agents moving toward lower energy regions. Note that the evolutionary trajectories include random perturbations (αε), meaning agents do not move straight toward the adjacent minimum.
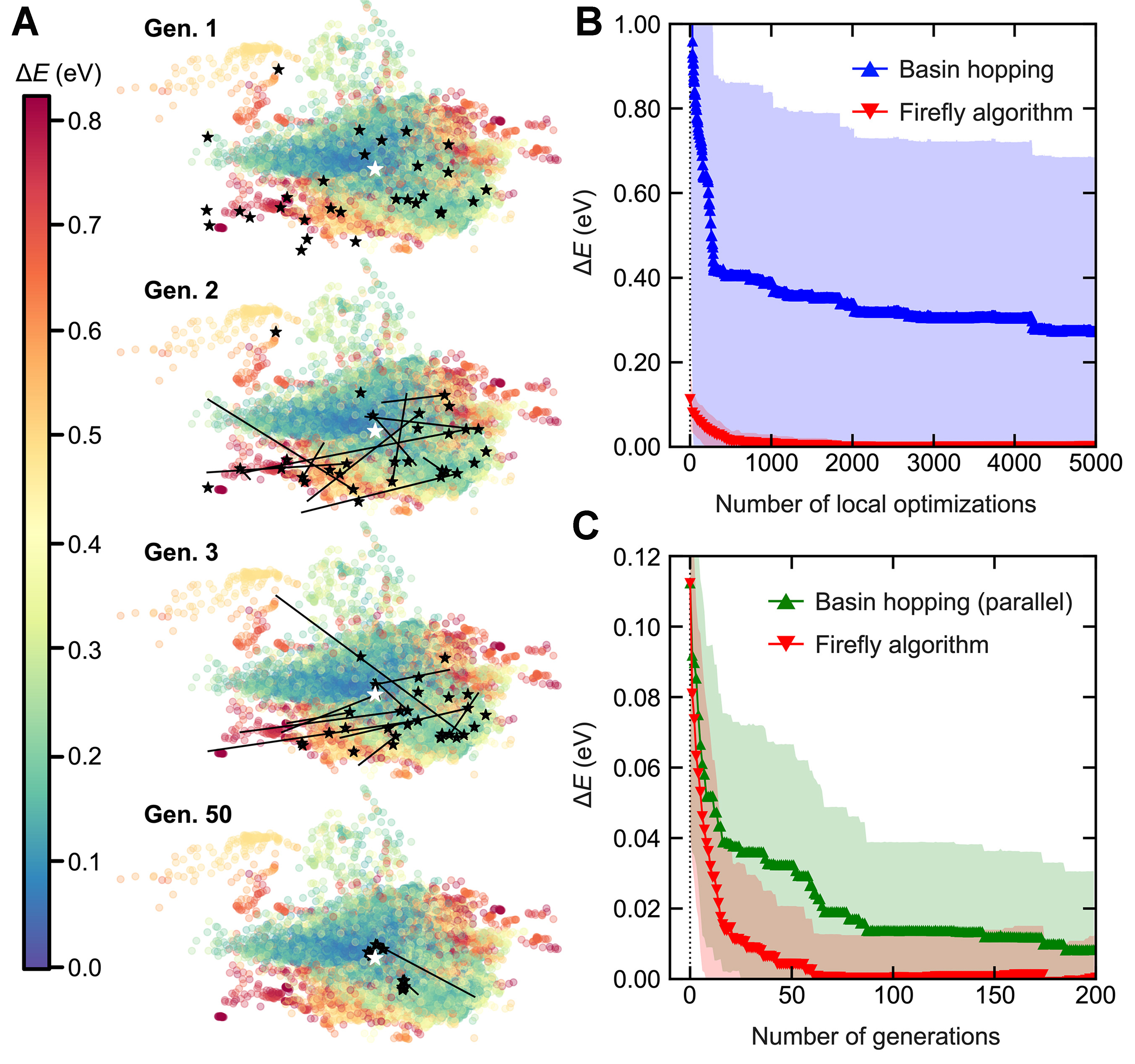
Figure 4. (A) Scatter plot on the PES generated by an attention-based VAE, colored according to relative energy compared to the global minimum. Black stars indicate agent positions in FA searches, with arrows tracing their evolutionary paths; (B) Performance comparison of 24 independent single BH runs (colored in blue) and 24 independent single FA searches (with 30 agents, colored in red) based on the number of local optimizations vs. energy difference from the global minimum; (C) Comparison between 24 independent FA and parallel BH runs. Both consist of 30 agents in each generation, and start from the same initial structures generated from FA runs. In (B) and (C), lines with markers indicate the mean and shaded regions indicate the standard deviation over 24 independent runs. PES: Potential energy surface; VAE: variational autoencoder; FA: firefly algorithm; BH:
To statistically evaluate the FA efficiency, we performed several independent FA and BH runs, comparing the mean and standard deviation of energies as a function of the number of local optimizations. Each BH step corresponds to a single local optimization, and the mean and standard deviation of 24 independent BH runs are plotted in blue markers and shaded regions in Figure 4B, respectively. In contrast, since each generation of FA search contains 30 local optimizations and the agent with the lowest energy is selected, the number of local optimizations is represented by multiplication of the number of generations by 30. The mean and standard deviation of 24 independent FA is plotted in Figure 4B as red markers and shaded regions, respectively. Since the results of FA are far below those of BH, this indicates that a single FA run will likely achieve more efficient global optimization than a single BH run, given the same number of local optimization calculations.
However, this direct comparison between a single BH run and a single FA run may be influenced by two underlying factors. One is indeed the efficiency of the FA algorithm, and the other is the advantage of using multiple agents simultaneously. Since the best agent was selected from the population in each generation, this likely reduced the influence of randomness. To remove all other factors than the algorithm’s efficiency, we utilized a parallelized BH method, simultaneously running 30 independent BH calculations per generation, similar to FA.
Through this setup, the modified BH run becomes technically equivalent to the FA when the inter-agent communication term (β) is removed and the Metropolis acceptance criterion is adopted. By initializing each BH run with the same random structures generated from the FA runs, we effectively eliminated all factors other than inter-agent communication (or mating). A total of 24 independent parallel-BH runs were conducted, resulting in 720 independent BH runs overall. As shown in Figure 4C, although the parallel-BH results (shown in green) begin from the same starting point at generation 0, they exhibit broader standard deviations (wider shaded regions) and slower convergence (higher-positioned markers) compared to the FA. This strongly indicates that inter-agent communication within the FA significantly enhances robustness in global optimization, consistent with previous findings[53], and highlights the advantage of such communication (or mating) in evolutionary algorithms over single-agent stochastic global optimization methods such as BH.
Application: gold cluster
The previously discussed “8” surface oxide on Cu(111) system exhibited a relatively simple PES, lacking clearly distinguishable local minima apart from the global minimum. Therefore, to highlight the advantages of the FA algorithm, we selected the gold cluster (nAu = 11) system as the target system, which has been systematically analyzed by Goldsmith et al.[26]. This system serves as an excellent example demonstrating how the choice of target potential can significantly affect the predicted global minimum structure. While the global minimum predicted using the PBE exchange-correlation (xc) functional is a planar edge-capped elongated hexagon structure, the vdW or MBD corrected xc functional displayed a structurally distinct global minimum, with a non-planar trigonal prism structure, which is also supported by experimental results. In this case, even with the use of state-of-the-art active learning combined with highly efficient and robust global optimization algorithms, relying solely on the PBE xc functional as the target potential leads to the identification of only the planar structure. In fact, the more efficient the global optimization becomes, the more likely it is to overlook the non-planar configuration under a fixed target potential. To address this limitation, one could either adopt a more accurate target potential, such as a vdW-corrected functional [e.g., hybrid Heyd-Scuseria-Ernzerhof (HSE) functional including MBD effects[26]], or perform multiple global optimization runs to statistically sample metastable configurations. However, since the FA algorithm is inherently designed to identify multimodal solutions beyond the global minimum, it is expected to capture both planar and non-planar configurations even with the PBE xc functional in a single run, as schematically illustrated in Figure 5A.
To validate this expectation, we applied NARA to the gold cluster (nAu = 11) system using an active learning-based global optimization scheme. For constructing the initial dataset, we set ninit = 20 for single-point calculations and nforce_init = 2 for force evaluations to configure our first GNN model (GNN1). During the FA searches, NARA called the 13 additional short local optimization calculations based on target potential (i.e., DFT with PBE xc), based on the LCB criterion. The GNN architecture employed irreducible representations of “32 × 0e + 32 × 0o + 32 × 1e + 32 × 1o”, 3 message passing blocks, and a radius cutoff 5 Å. The FA parameters were set to α = 0.5, β0 = 0.9, γ = 0.05. Compared to “8” surface oxide on Cu(111) case, a larger γ was used to strengthen the distance decay in the attractiveness term, making the interactions more short-ranged. The optimization terminated at Generation 241 when the global minimum remained unchanged for the predefined maximum patience, Pmax = 75. The agents from the final generation were overlaid on the PES created from all previously encountered gold cluster structures, as illustrated in Figure 5B. Interestingly, several distinct structures were found, including both the planar structure (global minimum when using PBE xc) and the non-planar structure (global minimum when including vdW corrections), demonstrating the robustness and insensitivity of the FA-based NARA algorithm to target potentials.
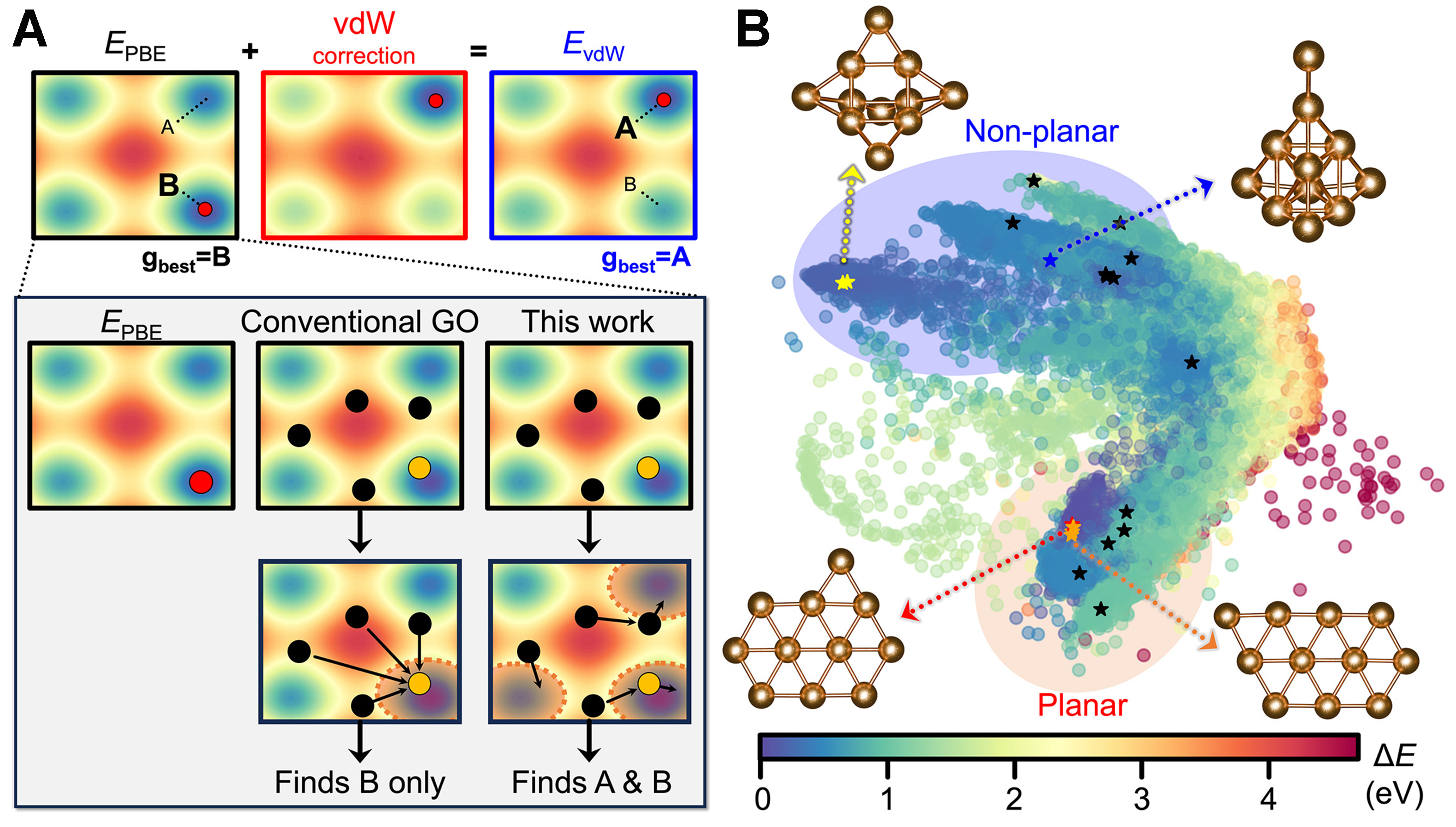
Figure 5. (A) Schematic illustration showing robustness to error in the target potential. In EPBE, Structure B represents the global minimum, but with vdW corrections, Structure A becomes more stable. Conventional GO algorithms always tend toward Structure B, the minimum of the chosen target potential, whereas the FA explores both minima, capturing both A and B (shaded region); (B) Agents from the final generation from a single NARA active learning run are shown on the PES. Top-left structure: non-planar global minimum (capped trigonal prism). Bottom-left structure: planar global minimum (edge-capped elongated hexagon). vdW: Van der Waals; GO: global optimization; FA: firefly algorithm; NARA: nature-inspired algorithm for robust atomic structure search; PES: potential energy surface; PBE: Perdew, Burke, and Ernzerhof.
CONCLUSION
In this study, we proposed NARA, a robust on-the-fly global optimization framework based on the FA. Compared to previous FA-based approaches, NARA significantly accelerates structural evolution by incorporating a machine learning-based energy and force prediction module. Additionally, by introducing a symmetry-invariant local descriptor-based atom index matching step, we efficiently addressed symmetry operations in the structure evolution process. The performance of our framework was validated through two case studies: the “8” surface oxide on Cu(111), showcasing superior global optimization performance, and the gold cluster system, demonstrating NARA’s ability to deliver multimodal solutions. Our approach notably enhances robustness by (i) integrating a GNN model with learning-loss-based rapid and accurate uncertainty quantification; and (ii) adopting the advantage of FA: attenuating structural attractiveness (-E) based on inter-structure distances, making it less sensitive to errors in the chosen target potential as illustrated in Figure 5A. NARA thus provides a novel, efficient approach for identifying energetically competitive structures within a single optimization run, representing a significant milestone applicable broadly to both non-periodic and periodic systems.
Building on these advancements, we outline several directions for further refinement. Future work may extend the framework to open systems and variable-cell optimizations, which are relevant to broader classes of atomistic structure searches. Although we examined scalability on a larger copper surface oxide system [Supplementary Figure 6], achieving consistent performance across diverse system sizes and landscape characteristics would benefit from systematic benchmarking and hyperparameter optimization. Furthermore, the effectiveness of FA-driven communication and symmetry-invariant index matching can vary with the degree of structural order, and additional validation across a wider range of systems would be beneficial. Finally, beyond the current global-best patience rule, a more robust convergence assessment would be advantageous by tracking the stabilization of multiple low-energy basins, for example, via structural clustering or the persistence of the top-k candidate set. These developments can be incorporated naturally within the present NARA workflow, since the GNN-based energy and force prediction, learning loss-based uncertainty quantification, and FA-based search are implemented as separable components, and this modularity supports applicability to both periodic and non-periodic systems.
DECLARATIONS
Author’s contributions
Software, investigation, writing - original draft: Lee, G.
Supervision, conceptualization, writing - review and editing: Stampfl, C.; Soon, A.
Availability of data and materials
Source code of nature-inspired algorithm for robust atomic structure search (NARA) is available at https://github.com/materials-theory/NARA, while LLUMYS is available at https://github.com/materials-theory/LLUMYS.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (RS-2023-NR077023, RS-2024-00468995) and by the Australian Research Council (FL230100176). Computational resources have been kindly provided by the KISTI Supercomputing Center (KSC-2023-CRE-0127) and the Australian National Computational Infrastructure (NCI). We also acknowledge support from the Sydney Global Engagement Scheme: University of Sydney-Yonsei University Partnership Collaboration Award (PCA).
Conflicts of interest
Soon, A. is an Associate Editor of the journal AI Agent and was not involved in any steps of the editorial process, including reviewer’s selection, manuscript handling, or decision-making. The other authors declare that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Lee, Y.; Ly, T. T.; Lee, T.; et al. Completing the picture of initial oxidation on copper. Appl. Surf. Sci. 2021, 562, 150148.
2. Lee, G.; Lee, Y.; Palotás, K.; Lee, T.; Soon, A. Atomic structure and work function modulations in two-dimensional ultrathin CuI films on Cu(111) from first-principles calculations. J. Phys. Chem. C. 2020, 124, 16362-70.
3. Lee, S.; Lee, Y. J.; Lee, G.; Soon, A. Activated chemical bonds in nanoporous and amorphous iridium oxides favor low overpotential for oxygen evolution reaction. Nat. Commun. 2022, 13, 3171.
5. Wu, Y. A.; Mcnulty, I.; Liu, C.; et al. Facet-dependent active sites of a single Cu2O particle photocatalyst for CO2 reduction to methanol. Nat. Energy. 2019, 4, 957-68.
6. Wang, W.; Ning, H.; Fei, X.; et al. Trace ionic liquid-assisted orientational growth of Cu2 O (110) facets promote CO2 electroreduction to C2 products. ChemSusChem 2023, 16, e202300418.
7. Chen, H.; Fan, T.; Ji, Y. CO2 reduction mechanism on the Cu2 O(110) surface: a first-principles study. Chemphyschem 2023, 24, e202300047.
8. Eilert, A.; Cavalca, F.; Roberts, F. S.; et al. Subsurface oxygen in oxide-derived copper electrocatalysts for carbon dioxide reduction. J. Phys. Chem. Lett. 2017, 8, 285-90.
9. Favaro, M.; Xiao, H.; Cheng, T.; Goddard, W. A. 3rd; Yano, J.; Crumlin, E. J. Subsurface oxide plays a critical role in CO2 activation by Cu(111) surfaces to form chemisorbed CO2, the first step in reduction of CO2. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 6706-11.
10. Fields, M.; Hong, X.; Nørskov, J. K.; Chan, K. Role of subsurface oxygen on Cu surfaces for CO2 electrochemical reduction. J. Phys. Chem. C. 2018, 122, 16209-15.
11. Zhu, B.; Huang, W.; Lin, H.; et al. Vacancy ordering in ultrathin copper oxide films on Cu(111). J. Am. Chem. Soc. 2024, 146, 15887-96.
12. Chen, D.; Chen, L.; Zhao, Q.; Yang, Z.; Shang, C.; Liu, Z. Square-pyramidal subsurface oxygen [Ag4OAg] drives selective ethene epoxidation on silver. Nat. Catal. 2024, 7, 536-45.
13. Vilhelmsen, L. B.; Hammer, B. A genetic algorithm for first principles global structure optimization of supported nano structures. J. Chem. Phys. 2014, 141, 044711.
14. Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. CALYPSO: a method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063-70.
15. Glass, C. W.; Oganov, A. R.; Hansen, N. USPEX - evolutionary crystal structure prediction. Comput. Phys. Commun. 2006, 175, 713-20.
16. Kim, H. J.; Lee, G.; Oh, S. V.; Stampfl, C.; Soon, A. Recalibrating the experimentally derived structure of the metastable surface oxide on copper via machine learning-accelerated in silico global optimization. ACS. Nano. 2024, 18, 4559-69.
17. Jung, H.; Sauerland, L.; Stocker, S.; Reuter, K.; Margraf, J. T. Machine-learning driven global optimization of surface adsorbate geometries. npj. Comput. Mater. 2023, 9, 114.
18. Lee, Y.; Lee, T. Machine-learning-accelerated surface exploration of reconstructed BiVO4(010) and characterization of their aqueous interfaces. J. Am. Chem. Soc. 2025, 147, 7799-808.
19. Tong, Q.; Xue, L.; Lv, J.; Wang, Y.; Ma, Y. Accelerating CALYPSO structure prediction by data-driven learning of a potential energy surface. Faraday. Discuss. 2018, 211, 31-43.
20. Bisbo, M. K.; Hammer, B. Global optimization of atomic structure enhanced by machine learning. Phys. Rev. B. 2022, 105, 245404.
21. Merte, L. R.; Bisbo, M. K.; Sokolović, I.; et al. Structure of an ultrathin oxide on Pt3Sn(111) solved by machine learning enhanced global optimization. Angew. Chem. Int. Ed. Engl. 2022, 61, e202204244.
22. Rønne, N.; Christiansen, M. V.; Slavensky, A. M.; et al. Atomistic structure search using local surrogate model. J. Chem. Phys. 2022, 157, 174115.
23. Kaappa, S.; Del Río, E. G.; Jacobsen, K. W. Global optimization of atomic structures with gradient-enhanced Gaussian process regression. Phys. Rev. B. 2021, 103, 174114.
24. Kaappa, S.; Larsen, C.; Jacobsen, K. W. Atomic structure optimization with machine-learning enabled interpolation between chemical elements. Phys. Rev. Lett. 2021, 127, 166001.
25. Larsen, C.; Kaappa, S.; Vishart, A. L.; Bligaard, T.; Jacobsen, K. W. Machine-learning-enabled optimization of atomic structures using atoms with fractional existence. Phys. Rev. B. 2023, 107, 214101.
26. Goldsmith, B. R.; Florian, J.; Liu, J.; et al. Two-to-three dimensional transition in neutral gold clusters: the crucial role of van der Waals interactions and temperature. Phys. Rev. Mater. 2019, 3, 016002.
27. Kruglov, I. A.; Yanilkin, A. V.; Propad, Y.; Mazitov, A. B.; Rachitskii, P.; Oganov, A. R. Crystal structure prediction at finite temperatures. npj. Comput. Mater. 2023, 9, 197.
28. Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B. 1993, 47, 558.
29. Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. Condens. Matter. 1996, 54, 11169-86.
30. Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999, 59, 1758.
31. Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865-8.
32. Chen, X.; Zhao, Y.; Wang, L.; Li, J. Recent progresses of global minimum searches of nanoclusters with a constrained Basin-Hopping algorithm in the TGMin program. Comput. Theor. Chem. 2017, 1107, 57-65.
33. Peterson, A. A. Global optimization of adsorbate–surface structures while preserving molecular identity. Top. Catal. 2013, 57, 40-53.
34. Hjorth Larsen, A.; Jørgen Mortensen, J.; Blomqvist, J.; et al. The atomic simulation environment-a Python library for working with atoms. J. Phys. Condens. Matter. 2017, 29, 273002.
35. Kingma, D. P. Welling, M.. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114. Available online: https://doi.org/10.48550/arXiv.1312.6114. (accessed 30 Mar 2026).
36. Diederik, P. K.; Max, W. An introduction to variational autoencoders. arXiv 2019, arXiv:1906.02691. Available online: https://doi.org/10.48550/arXiv.1906.02691. (accessed 30 Mar 2026).
37. Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Liò, P.; Bengio, Y. Graph attention networks. arXiv 2017, arXiv:1710.10903. Available online: https://doi.org/10.48550/arXiv.1710.10903. (accessed 30 Mar 2026).
38. Yang, X. S. Firefly algorithms for multimodal optimization. In Stochastic Algorithms: Foundations and Applications; Watanabe, O., Zeugmann, T., Eds.; Lecture Notes in Computer Science, Vol. 5792; Springer Berlin Heidelberg, 2009; pp 169-78.
39. Togo, A.; Shinohara, K.; Tanaka, I. Spglib: a software library for crystal symmetry search. Sci. Technol. Adv. Mater. Methods. 2024, 4, 2384822.
41. Batzner, S.; Musaelian, A.; Sun, L.; et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 2022, 13, 2453.
42. Yoo, D.; Kweon, I. S. Learning loss for active learning. In 2019 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), Long Beach, USA, June 15-20, 2019; IEEE, 2019; pp. 93-102.
43. Avendaño-Franco, G.; Romero, A. H. Firefly algorithm for structural search. J. Chem. Theory. Comput. 2016, 12, 3416-28.
44. Mitra, A.; Jana, G.; Agrawal, P.; Sural, S.; Chattaraj, P. K. Integrating firefly algorithm with density functional theory for global optimization of Al42- clusters. Theor. Chem. Acc. 2020, 139, 32.
45. Schran, C.; Thiemann, F. L.; Rowe, P.; Müller, E. A.; Marsalek, O.; Michaelides, A. Machine learning potentials for complex aqueous systems made simple. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2110077118.
46. Tokita, A. M.; Behler, J. How to train a neural network potential. J. Chem. Phys. 2023, 159, 121501.
47. Kang, K.; Purcell, T. A. R.; Carbogno, C.; Scheffler, M. Accelerating the training and improving the reliability of machine-learned interatomic potentials for strongly anharmonic materials through active learning. Phys. Rev. Mater. 2025, 9, 063801.
48. Tan, A. R.; Urata, S.; Goldman, S.; Dietschreit, J. C. B.; Gómez-bombarelli, R. Single-model uncertainty quantification in neural network potentials does not consistently outperform model ensembles. npj. Comput. Mater. 2023, 9, 225.
49. Kabsch, W. A solution for the best rotation to relate two sets of vectors. Acta. Cryst. A. 1976, 32, 922-3.
50. Xie, Y.; Vandermause, J.; Sun, L.; Cepellotti, A.; Kozinsky, B. Bayesian force fields from active learning for simulation of inter-dimensional transformation of stanene. npj. Comput. Mater. 2021, 7, 40.
51. Christiansen, M. V.; Rønne, N.; Hammer, B. Efficient ensemble uncertainty estimation in Gaussian processes regression. Mach. Learn. Sci. Technol. 2024, 5, 045029.
52. Bisbo, M. K.; Hammer, B. Efficient global structure optimization with a machine-learned surrogate model. Phys. Rev. Lett. 2020, 124, 086102.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].