

A closed-loop universal catalyst design workflow ready for AI agents
 0
0 
MAIN TEXT
The search for high-performance electrocatalysts is increasingly limited not by the lack of candidate materials but by the lack of efficient workflows that can connect data, prediction, validation, and iterative design. In this context, the recent work by Wei, Li, Yang, and coauthors reports a timely and important advance[1]: a universal catalyst design framework for the two-electron water oxidation reaction (2e- WOR)[2] toward electrochemical hydrogen peroxide (H2O2) synthesis. Rather than presenting another isolated machine-learning (ML) model, this study combines descriptor engineering, database integration, microkinetic analysis, and experimental validation into a transferable discovery workflow. Most importantly, it also points toward a natural next step: the future integration of such a workflow into AI Agent systems that can orchestrate the full research loop.
H2O2 is an attractive energy-relevant chemical, and its electrochemical production through 2e- pathways offers a potentially cleaner and more distributed alternative to conventional synthesis[3]. Yet, catalyst design for 2e- WOR remains challenging. Previous studies have often relied on thermodynamic limiting-potential models[4], which are useful for intuition but often fail to capture experimentally relevant activity trends. At the same time, most ML descriptors used in catalysis remain family-specific, limiting their transferability across diverse material classes such as alloys, oxides, perovskites, and single-atom catalysts. This work addresses both challenges simultaneously: it develops the weighted atom-centered symmetry function (wACSF) descriptors to unify geometric and chemical information at active sites, and couples these descriptors to precise microkinetic modelling rather than stopping at static adsorption-energy regression. Unlike conventional atom-centered symmetry function (ACSF)-based approaches, which capture only geometric features of atomic local environments, the dual representation of the proposed wACSF descriptors allows for a more physically meaningful description of catalytic activity and improves transferability across different classes of materials. By employing the same predefined symmetry-function parameters for different systems, wACSF maintains a consistent descriptor dimensionality and demonstrates universality across diverse catalyst families.
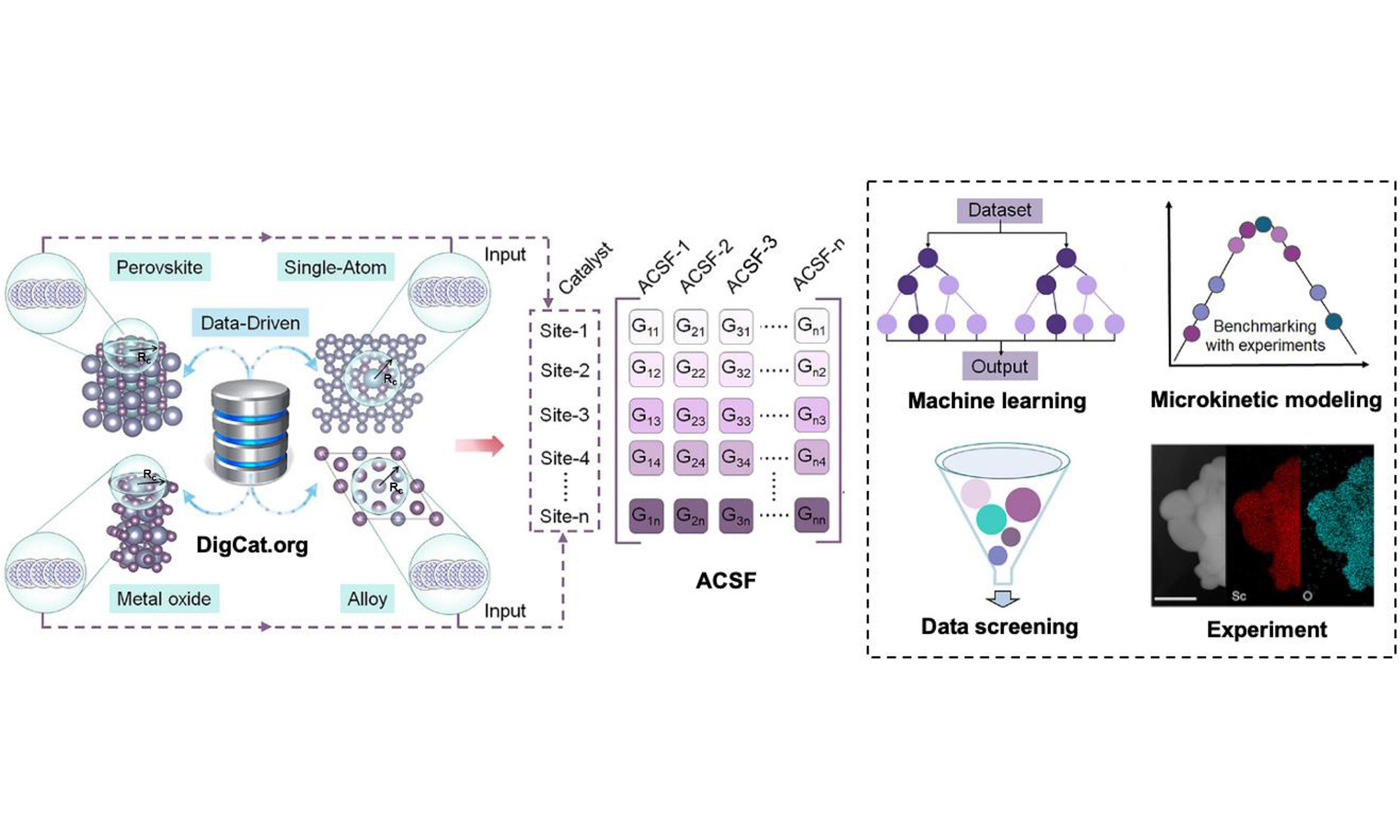
A major strength of the paper is its explicit workflow design. As summarized in Figure 1, the framework begins with feature extraction from the Digital Catalysis Platform (DigCat: www.digcat.org)[5], followed by descriptor generation, feature selection, ML training, benchmarking against experimental data, and finally ML-accelerated catalyst screening. This is not just a predictive model, but a structured and transferable research pipeline. The authors compiled a dataset of 962 materials spanning intermetallic alloys, metal oxides, perovskites, and single-atom catalysts, and used a consistent, predefined symmetry-function parameterization across all classes. This consistency is important: the model does not merely interpolate within one single catalyst family, but instead learns a representation intended to transfer across chemically distinct systems. The resulting eXtreme Gradient Boosting (XGBoost) models achieved strong predictive performance for both ΔGOH* and ΔGO*, providing a practical basis for large-scale screening.
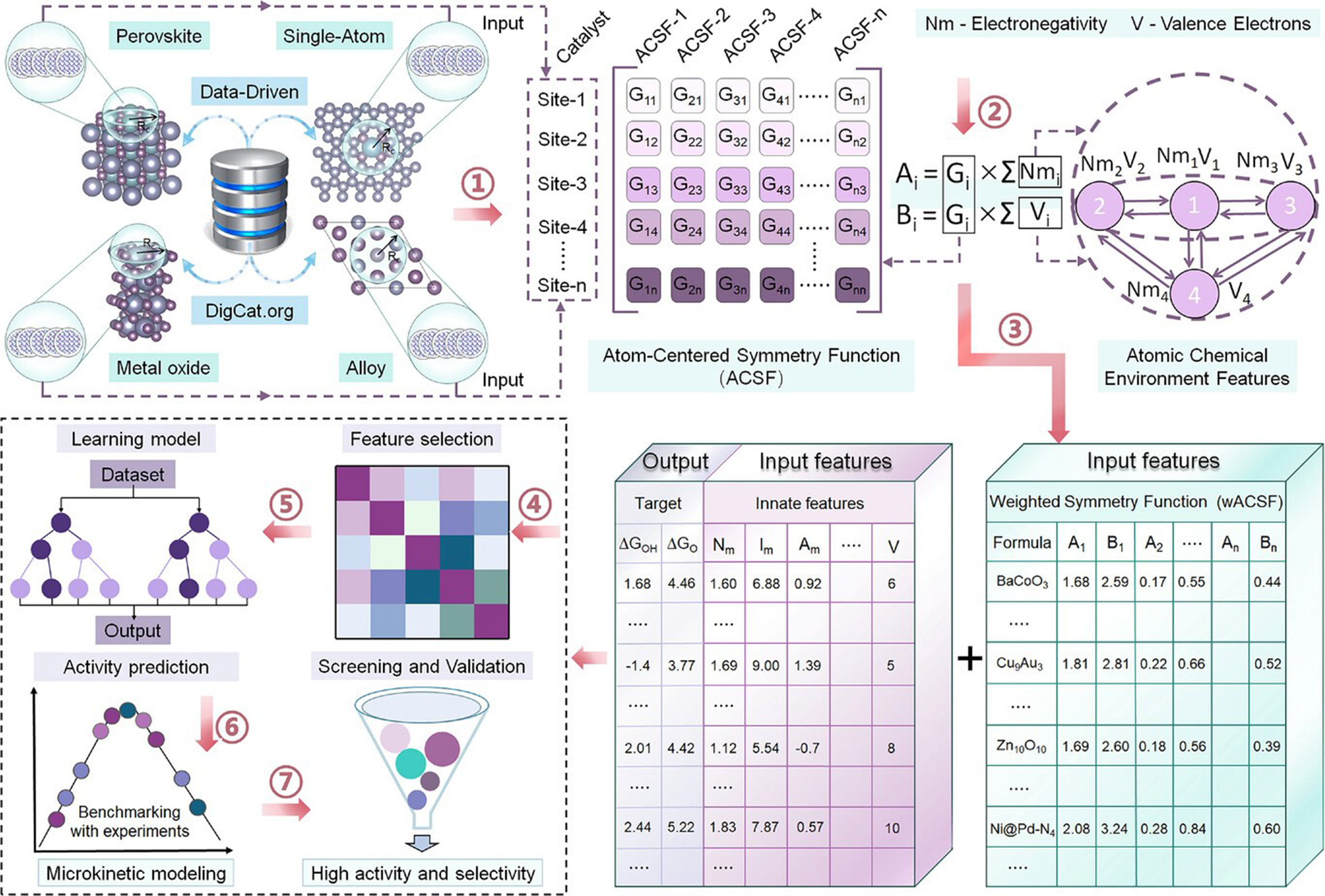
Figure 1. Overview of the universal catalyst design workflow integrating DigCat-based data extraction, wACSF descriptor construction, machine learning, microkinetic benchmarking, and catalyst screening. This figure is quoted with permission from Wei, Li, Yang et al.[1]. DigCat: Digital Catalysis Platform; wACSF: weighted atom-centered symmetry function.
What makes this paper especially notable is that it does not treat adsorption-energy prediction as the endpoint. Instead, the predicted descriptors are fed into a microkinetic volcano framework. As shown in Figure 2, the authors establish a new microkinetic volcano for 2e- WOR, compare it with the conventional thermodynamic volcano, and then use the descriptor relationships together with selectivity constraints to filter catalyst candidates. This approach matters because microkinetic modelling provides a more realistic bridge between theory and experiment than a purely thermodynamic screening strategy. The resulting volcano reproduces reported activity trends well, with the apex centered around ΔGOH* ≈ 1.76 eV, and enables a rational selection of candidates that satisfy both activity and selectivity requirements for H2O2 production. The screening logic is therefore mechanistically informed rather than purely statistical.
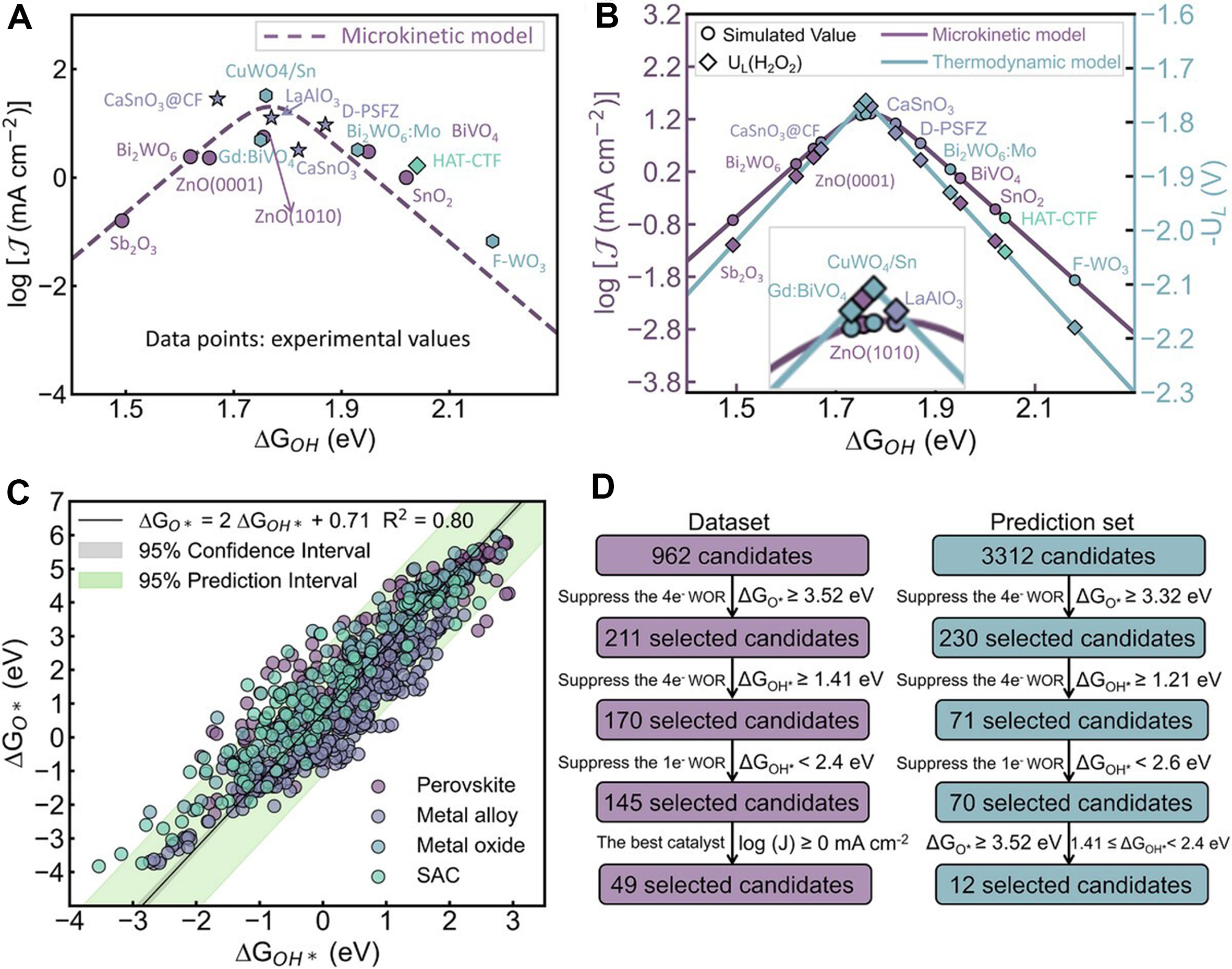
Figure 2. Microkinetic volcano analysis and screening logic for 2e- WOR, showing how activity, selectivity, and descriptor scaling relations jointly define promising catalyst space. This figure is quoted with permission from Wei, Li, Yang et al.[1]. D-PSFZ: Pr1.0Sr1.0Fe0.75Zn0.25O4-δ; R2: coefficient of determination.
The workflow then advances from retrospective validation to prospective discovery. After screening, the model identifies a small set of promising catalysts, among which LiScO2 emerges as the top candidate. This is where the study becomes particularly convincing. As shown in Figure 3, the authors provide comprehensive experimental validation, including structural characterization, electrochemical measurements, comparison between predicted and observed performance, and long-term stability. LiScO2 achieves about 90% Faradaic efficiency for H2O2 at 2.2 V vs. reversible hydrogen electrode (RHE), maintains over 80% selectivity up to
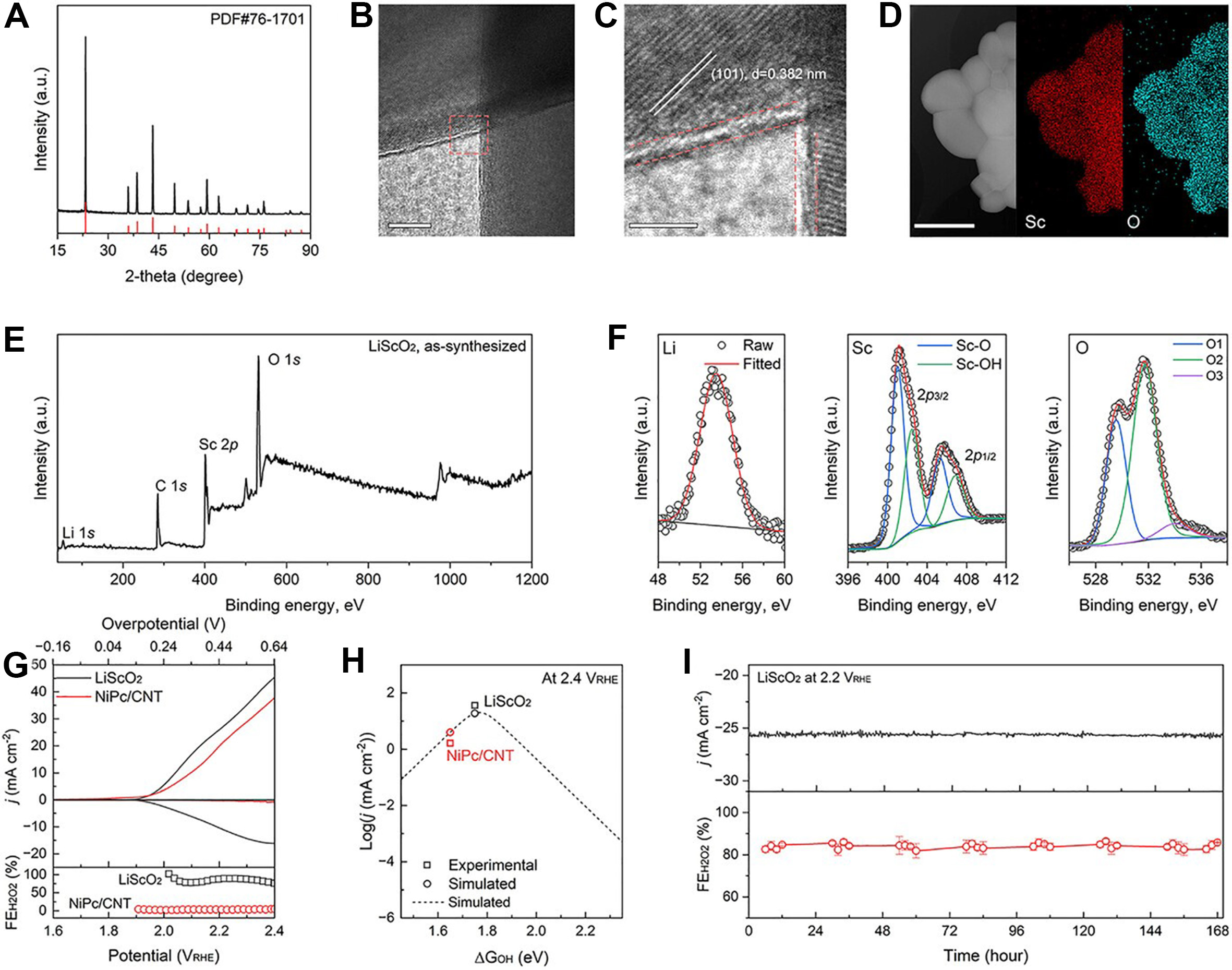
Figure 3. Experimental validation of the top-predicted LiScO2 catalyst, including structural characterization, electrochemical H2O2 performance, theory-experiment comparison, and long-term stability. This figure is quoted with permission from Wei, Li, Yang et al.[1]. PDF: Powder Diffraction File; NiPc: nickel (II) phthalocyanine; CNT: carbon nanotube; VRHE: voltage versus reversible hydrogen electrode.
Beyond the immediate catalytic result, the broader significance of this work lies in its architecture. The paper already describes a unified design framework integrating database automation, ML, new microkinetic modelling, and cross-material screening, and highlights its extendability to other reactions such as oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), which share key descriptors GOH* and GO*[6,7]. That statement is crucial. It implies that the value of this work is not limited to one single catalyst or reaction, but instead provides a modular template for future AI-enabled research infrastructure. In other words, the central product of the study is not only the newly discovered high-performance LiScO2 catalyst, but a reusable discovery protocol.
This is precisely where AI Agents enter the picture[8]. At present, the reported workflow is highly structured but still largely executed as a research pipeline assembled by human researchers. In the near future, however, each stage could be naturally assigned to specialized agents: one for literature and database retrieval, one for descriptor generation and quality control, one for ML model training and uncertainty analysis, one for microkinetic simulation, one for candidate prioritization under multi-objective constraints, and one for linking predictions to experimental planning. Under such a framework, AI Agents would not replace the physical and theoretical rigour of the present study; rather, they would operationalize it. The wACSF-based workflow provides a concrete chain for agents to orchestrate: database → descriptor → model → microkinetics → candidate selection → experiment → feedback.
This perspective is important because one of the central challenges in AI for science today is moving from conversational intelligence to executable research systems. Many AI demonstrations remain trapped at the stage of summarization, suggestion, or isolated prediction. By contrast, the framework reported here already contains the ingredients of a deployable agentic workflow. Figure 1 can be interpreted not only as a computational pipeline, but also as a proto-agent architecture. Figure 2 defines the decision layer, where kinetic and selectivity constraints determine which candidates move forward. Figure 3 closes the loop by showing how predictions are validated experimentally and can, in principle, be fed back into the system for model refinement. Together, these figures show a workflow that is not merely compatible with AI Agents but ready to be upgraded by them.
Looking ahead, several opportunities arise from this direction. First, agentic integration could improve throughput by automating the repeated transitions between database query, descriptor computation, microkinetic evaluation, and candidate ranking. Second, robustness could be improved by enforcing standardized benchmarking and uncertainty-aware decision-making before expensive experiments are launched. Third, transferability could be enhanced by adapting the same workflow skeleton to related electrocatalytic reactions. The paper already points to ORR and OER as natural extensions, since ΔGOH* and ΔGO* remain broadly informative reactivity descriptors in those systems. If these modules are embedded into AI Agent frameworks, future platforms may not only screen catalysts but also autonomously decide which theory calculations to run next, which literature gaps to fill, and which experiments are most valuable for updating the model. In this context, and given a few studies on autonomous experimentation[9], and active learning frameworks applied to ORR and OER[10], this development is particularly valuable.
In this sense, the present study marks more than a technical improvement in catalyst screening. It illustrates a deeper shift in how catalyst discovery can be organized. The combination of transferable descriptors, mechanistically grounded screening, and experimentally verified prediction provides a solid foundation for the next generation of agent-driven research systems. As the field moves beyond isolated AI models, workflows like this one may become the real units of progress. The future of AI in catalysis will depend not only on better models but on better orchestration of models, databases, theory, and experiments. This work offers a strong example of that future.
DECLARATIONS
Authors’ contributions
Made substantial contributions to conception and design of the study and performed data analysis and interpretation: Chen, L.; Zhan, S.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Grant, Formas - a Swedish Research Council for Sustainable Development, Sweden (N 2023-01559) and the Åforsk foundation (nr 25-499).
Conflicts of interest
All authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Liu, Z.; Liu, Y.; Zhang, Y.; et al. Universal catalyst design framework for electrochemical hydrogen peroxide synthesis facilitated by local atomic environment descriptors. Angew. Chem. Int. Ed. 2025, 65, e18027.
2. Cai, S.; Lai, C.; Li, L.; Sun, H.; Ma, M. Toward dual-electrode full-cell production: a review on electrosynthesis of H2O2 via 2e- ORR and 2e- WOR. Adv. Sustain. Syst. 2026, 10, e01632.
3. Shi, X.; Siahrostami, S.; Li, G.; et al. Understanding activity trends in electrochemical water oxidation to form hydrogen peroxide. Nat. Commun. 2017, 8, 701.
4. Baek, J.; Jin, Q.; Johnson, N. S.; et al. Discovery of LaAlO3 as an efficient catalyst for two-electron water electrolysis towards hydrogen peroxide. Nat. Commun. 2022, 13, 7256.
5. Zhang, D.; Bao, Z.; Chu, Y.; et al. Digital Catalysis Platform (DigCat): a gateway to big data and AI-powered innovations in catalysis. ChemRxiv 2024. Available online: https://doi.org/10.26434/chemrxiv-2024-9lpb9 (accessed 26 May 2026).
6. Zhang, D.; Wang, Z.; Liu, F.; et al. Unraveling the pH-dependent oxygen reduction performance on single-atom catalysts: from single- to dual-sabatier optima. J. Am. Chem. Soc. 2024, 146, 3210-9.
7. Dickens, C. F.; Kirk, C.; Nørskov, J. K. Insights into the electrochemical oxygen evolution reaction with ab initio calculations and microkinetic modeling: beyond the limiting potential volcano. J. Phys. Chem. C. 2019, 123, 18960-77.
9. Seifrid, M.; Pollice, R.; Aguilar-granda, A.; et al. Autonomous chemical experiments: challenges and perspectives on establishing a self-driving lab. Acc. Chem. Res. 2022, 55, 2454-66.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].