


Single-cell sequencing in precision management of chronic myeloid leukemia
 0
0 Abstract
Single-cell sequencing technology provides a high-resolution perspective for the precision diagnosis and treatment of chronic myeloid leukemia (CML). Cellular and molecular heterogeneity in CML is closely associated with tyrosine kinase inhibitor resistance, residual disease, and disease progression. However, the averaged signals generated by conventional bulk sequencing limit the identification of minor resistant clones, state-dependent leukemia stem cell (LSC) programs, and complex microenvironmental interactions. This review summarizes frontier applications of single-cell RNA sequencing and related single-cell omics technologies in CML, including molecular mechanism analysis, tumor microenvironment profiling, resistance mechanism exploration, biomarker discovery, and the translation of these findings into risk stratification, treatment monitoring, and individualized therapeutic strategies. By integrating recent global research findings, this review discusses the value of single-cell technology in revealing the functional heterogeneity of LSCs and provides a balanced perspective on the future integration of spatial multi-omics, artificial intelligence-assisted analysis, and clinical decision systems. The aim is to clarify how CML diagnosis and treatment may move from empiric disease monitoring toward a more precise, single-cell-informed model while acknowledging current technical, economic, regulatory, and validation barriers.
Keywords
INTRODUCTION
Chronic myeloid leukemia (CML) is a quintessential myeloproliferative neoplasm, accounting for approximately 15% of adult leukemia cases[1]. As a malignant clonal disease originating from pluripotent hematopoietic stem cells (HSCs), its molecular pathology is rooted in the reciprocal translocation between the ABL1 gene on chromosome 9 and the BCR gene on chromosome 22, forming the Philadelphia chromosome[2,3]. The resulting BCR::ABL1 fusion gene serves as the primary driver of disease development[2,3]. The encoded BCR::ABL1 fusion protein possesses constitutive tyrosine kinase activity, triggering multiple downstream signaling pathways, including RAS/MAPK, PI3K/AKT/mTOR, JAK/STAT, and JUN kinase[4-8].
Recent proteogenomic integrative analyses have further suggested that the PI3K/AKT/mTOR signaling pathway may represent a therapeutically vulnerable program across myeloid malignancies and may help stratify the risk of leukemic transformation in broader myeloid contexts[9]. CD74-centered inflammatory signaling has also been reported in related myeloid disorders; however, its direct relevance to CML should be interpreted as contextual rather than CML-specific evidence[10]. In CML, BCR::ABL1-driven activation of proliferative and survival pathways disrupts the precise homeostasis of proliferation, differentiation, and apoptosis, granting myeloid progenitor cells increased proliferative capacity and the ability to evade programmed cell death, ultimately leading to the malignant expansion of myeloid cells in the bone marrow and peripheral blood[11].
Since the clinical introduction of the first-generation tyrosine kinase inhibitor (TKI), imatinib, in the early 21st century, the diagnostic and therapeutic paradigm of CML has been substantially changed. Contemporary long-term evidence indicates that the 10-year survival rate for patients with chronic-phase CML has improved from approximately 20% before effective TKI therapy to approximately 80%-90% in the modern TKI era, with CML-specific survival exceeding 90% in patients who achieve sustained molecular responses[1]. However, despite the advent of second-generation TKIs (dasatinib, nilotinib, bosutinib) and third-generation agents (ponatinib, olverembatinib), approximately 20% to 30% of patients still experience primary intolerance, suboptimal response, or secondary resistance[12-15]. Such treatment resistance is partly attributed to the inherent cellular heterogeneity of CML clones, which provides an important rationale for performing single-cell multi-omics analyses.
Single-cell multi-omics evidence suggests that this clinical divergence is partly rooted in baseline cellular heterogeneity; patients with poor deep molecular responses (DMR) may harbor specific primitive progenitor clusters characterized by high inflammatory signatures (e.g., TNF-alpha, NF-kappaB), complement-related programs, immune dysregulation, or metabolic abnormalities even prior to TKI initiation[16,17]. Furthermore, early-phase cytoreductive interventions, such as hydroxyurea (HU), have been shown at the single-cell level to alter the stress states and molecular characteristics of stem and progenitor cells, potentially influencing subsequent TKI sensitivity[18].
A central challenge of resistance and recurrence lies in the persistent existence of leukemia stem cells (LSCs). LSCs typically reside in a highly quiescent state with low metabolic rates and specialized molecular defense mechanisms, helping them evade TKI-induced apoptosis and remain a potential seed bank for relapse after long-term remission. Observations in related myeloid malignancies also provide contextual, non-CML-specific support for the broader concept that stemness maintenance, cooperating mutations, and inflammatory remodeling can shape clonal evolution[19]. Through single-cell sequencing, researchers have identified novel markers such as FBXO3; specifically, the FBXO3-DUSP9 axis may mediate LSC maintenance and TKI resistance by modulating MAPK signaling[20]. Historically, traditional bulk RNA-sequencing (bulk RNA-seq) has faced substantial limitations in resolving these issues. Because bulk profiling captures the averaged expression of thousands of cells, it can dilute signals from rare LSCs, confound malignant and non-malignant cell sources, and introduce technical or biological biases related to cellular composition, batch processing, and sample heterogeneity[21,22]. Single-cell methods address these limitations by assigning transcriptional, immunophenotypic, epigenetic, and mutational information to individual cells, thereby enabling the reconstruction of resistant clones and their niche interactions.
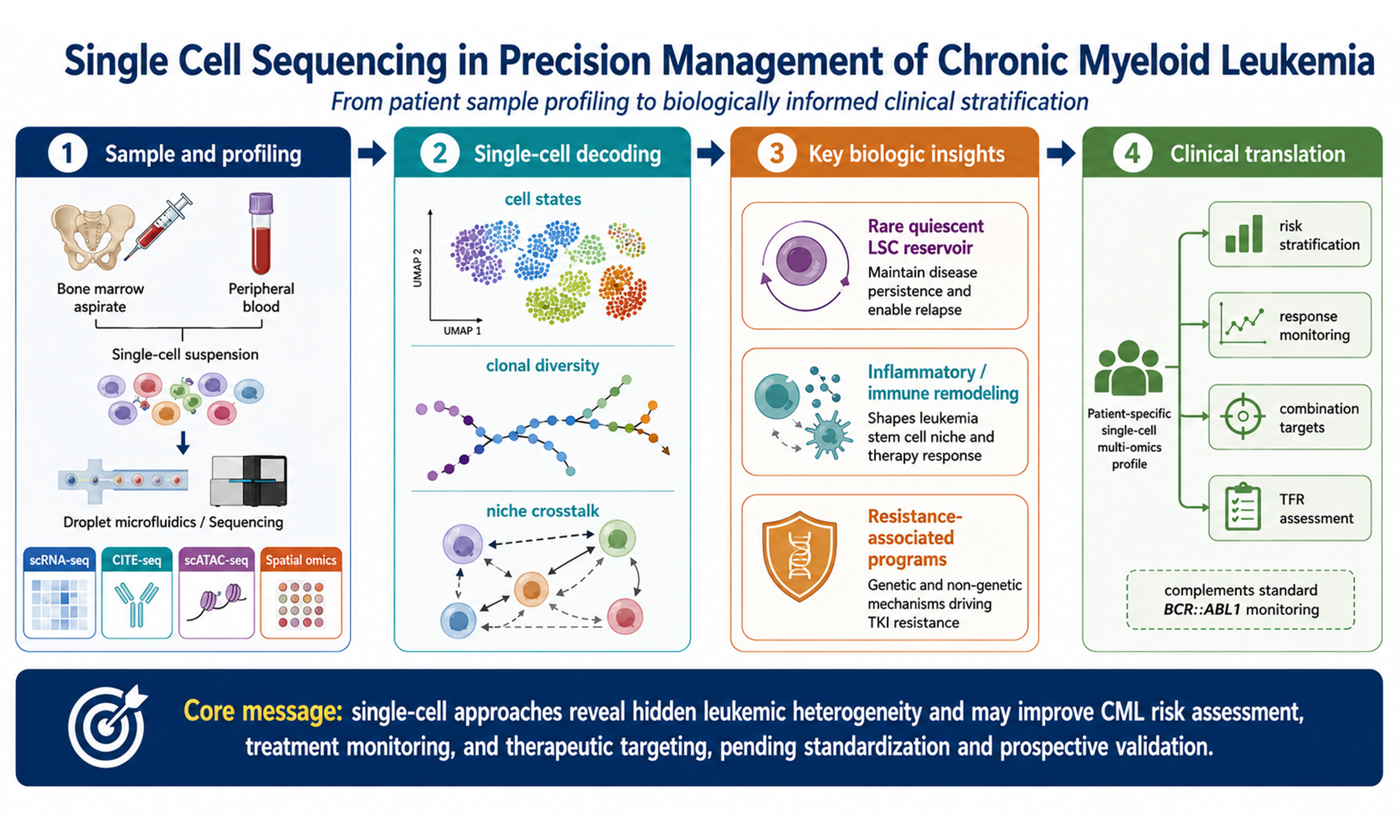
The rise of single-cell RNA sequencing (scRNA-seq) has helped overcome this resolution bottleneck by shifting the paradigm from population averaging to single-cell resolution[23,24]. First, scRNA-seq enables the deconstruction of the continuous evolution spectrum. Unlike the discrete stages defined by bulk analysis, longitudinal single-cell tracking suggests that CML progression may involve a continuous drift of transcriptional microstates, although the available longitudinal evidence remains partly dependent on preprint-level datasets and should therefore be interpreted as hypothesis-generating until replicated in peer-reviewed cohorts[25]. Second, it resolves genotype-phenotype linkage at the single-clone level. By jointly analyzing mutational and transcriptional landscapes, researchers can identify how specific sub-clones (e.g., those carrying NPM1 fusions or specific BCR::ABL1 mutations) acquire preferred transcriptional states that may contribute to drug resistance or extramedullary expansion[26,27]. Third, it uncovers the bystander effect within the niche. Single-cell atlases suggest that even BCR::ABL1-negative bystander cells (e.g., macrophages and T cells) can be transcriptionally reprogrammed by the leukemic environment to support leukemic cell survival and immune evasion[28,29]. The core genomic and signaling framework of CML pathogenesis is summarized in Figure 1. By analyzing LSC molecular features, heterogeneous microenvironment interactions, and clonal evolution at the single-cell level, this technology provides technical support and theoretical foundations for moving from pharmacological control toward measurable, cautiously defined single-cell-informed precision management.
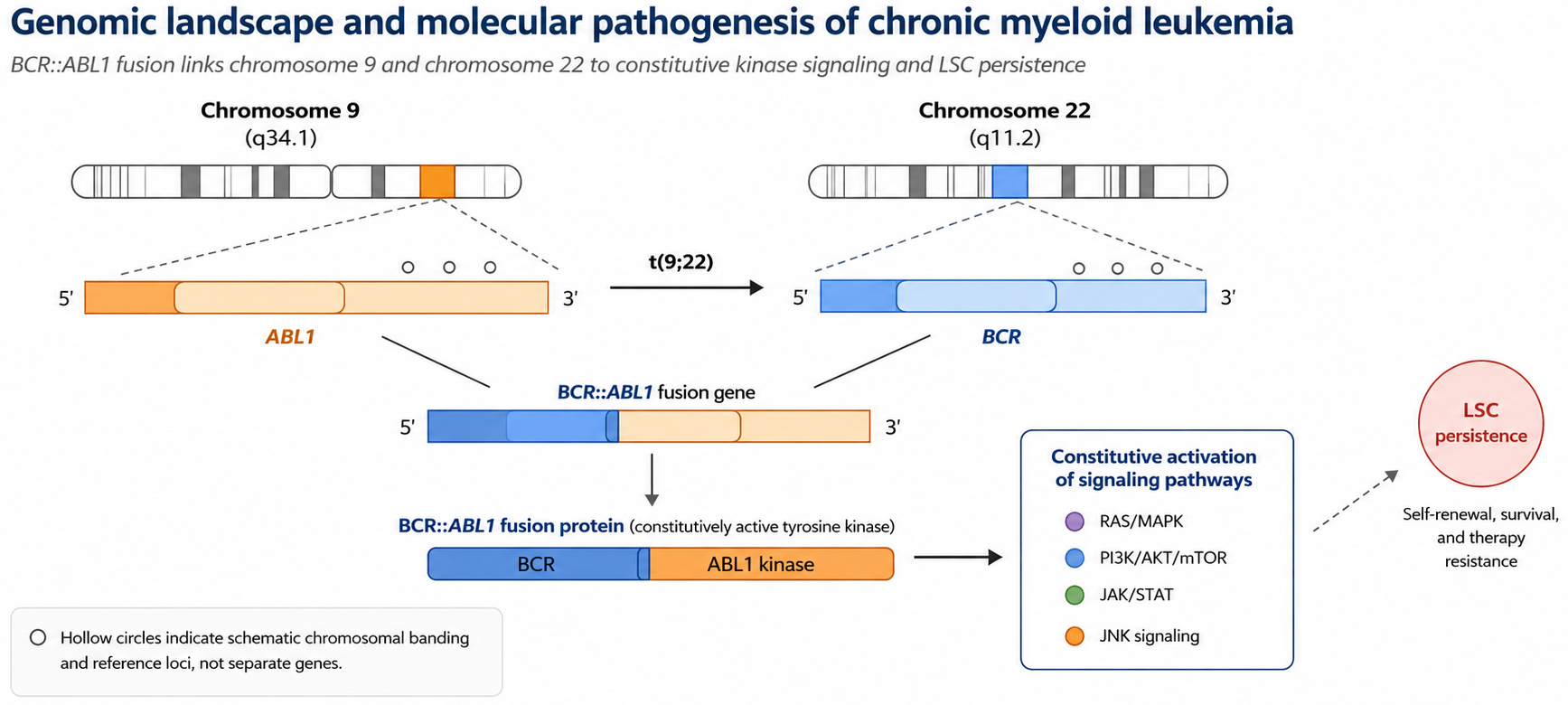
Figure 1. Genomic landscape and molecular pathogenesis of chronic myeloid leukemia. The primary molecular driver, the BCR::ABL1 fusion gene, originates from the reciprocal translocation between chromosomes 9 and 22 [t(9;22)(q34;q11)], resulting in the formation of the characteristic Philadelphia chromosome (Ph). This genomic aberration leads to constitutive activation of tyrosine kinase signaling, which serves as the foundational event in CML pathogenesis. The hollow circles on the orange chromosome schematically indicate chromosomal banding and reference loci used to orient the reader, rather than independent genes or mutations. This figure was redrawn by the authors using Python-based vector drawing and Microsoft Office-compatible elements; no third-party copyrighted materials were reused. CML: Chronic myeloid leukemia; Ph: philadelphia chromosome; LSC: leukemia stem cell.
TECHNICAL EVOLUTION, PLATFORM COMPARISON, AND LOGICAL SYSTEM OF SINGLE-CELL RNA SEQUENCING
Single-cell sequencing technology systems achieve multi-dimensional analysis of individual cell genomes, transcriptomes, and epigenomes by breaking the limitations of traditional population-based analysis[30-32]. As the earliest and most widely adopted technology, the core value of scRNA-seq in CML research lies in its capacity to directly identify and define rare cell subpopulations that are morphologically indistinguishable but molecularly distinct[33]. From early Smart-seq-based protocols to current droplet-based microfluidic platforms such as 10x Genomics, detection throughput has increased from hundreds of cells to tens of thousands of cells per experiment, enabling researchers to capture low-proportion LSCs amidst a vast background of normal hematopoietic cells[34-38].
Compared with traditional next-generation sequencing (NGS) based on population averages, single-cell technologies provide several critical optimizations. In multi-omics integration, CITE-seq enables parallel quantification of cell surface protein abundance and mRNA levels, allowing proteogenomic profiling to define stem and progenitor cell sub-clusters more precisely[16,39]. In genotype-phenotype linkage, targeted detection strategies can synchronously define BCR::ABL1 fusion status and other driver mutations, such as NPM1 fusions, at the single-cell level[26,27]. Finally, spatial transcriptomics and CODEX-based spatial proteomics enable direct observation of the spatial reconstruction of the bone marrow microenvironment and its relationship to leukemic evolution[40].
Critical comparison of major single-cell platforms
Smart-seq and its later derivatives provide relatively full-length transcript coverage and high sequencing depth per cell, making them valuable for splice isoform analysis, allele-level expression, and the detection of low-abundance transcripts. Their main limitations are lower throughput, higher per-cell cost, greater labor requirements, and reduced suitability for routine clinical screening when thousands of cells must be profiled from many patients[33-35]. In contrast, droplet-based platforms such as 10× Genomics offer substantially higher throughput, lower cost per cell, and standardized commercial workflows, which make them attractive for constructing CML atlases and screening heterogeneous diagnostic samples. However, droplet systems generally capture only a fraction of each transcript, have greater dropout rates, and may miss rare fusion transcripts or low-expression regulators unless paired with targeted enrichment or deep sequencing[36-38]. Therefore, Smart-seq-like approaches are better suited for mechanistic validation and transcript-level resolution, whereas 10× Genomics is more practical for large-scale cell-state mapping and exploratory patient stratification.
CITE-seq and related proteogenomic methods are particularly relevant to CML because potentially translatable LSC definitions depend heavily on surface markers such as CD26, CD25, CD93, and IL1RAP, while transcript abundance alone does not always predict surface protein abundance or signaling activity[16,39]. Their advantages include simultaneous immunophenotyping and transcriptomic profiling in the same cell, but they require validated antibody panels, careful compensation for antibody-derived tag background, and batch-standardized sample handling. scATAC-seq provides complementary information on chromatin accessibility and regulatory priming, enabling the identification of enhancers, transcription factor motifs, and epigenetic states linked to quiescence or blast crisis (BC) progression[30-32]. Its clinical limitations include sparse data matrices, more complex computational integration, and lower direct interpretability for clinicians compared with expression or flow cytometric readouts.
CyTOF and imaging mass cytometry provide high-dimensional protein measurement with broad antibody panels, enabling simultaneous analysis of immune subsets and kinase or phosphoprotein states. These methods are closer to established clinical immunophenotyping than sequencing-based assays, but they do not provide genome-wide transcriptomic or mutational information and require rigorous antibody validation. Spatial transcriptomics, CODEX, and three-dimensional imaging preserve tissue architecture and are therefore uniquely suited for studying the leukemic bone marrow niche, vascular disruption, macrophage remodeling, and localized immune exhaustion[40]. Their major constraints are higher cost, difficult tissue processing, limited throughput across large cohorts, and more challenging regulatory standardization. Thus, these platforms should be viewed not as interchangeable technologies but as tools selected according to the biological question, sample availability, depth requirement, and translational endpoint.
Biological differences across chronic phase, accelerated phase, and BC CML
The biological landscape captured by single-cell technologies differs across chronic phase, accelerated phase, and BC CML. In chronic phase, the dominant challenge is not massive loss of differentiation but the coexistence of normal HSCs, BCR::ABL1-positive stem and progenitor cells, and transcriptionally diverse LSC states with distinct growth rates and TKI sensitivity[16,39]. In this setting, single-cell analysis is most useful for identifying baseline cell-state composition, primitive quiescent clusters, inflammatory signatures, and immune-cell features that may influence DMR, treatment persistence, and potential future treatment-free remission. During accelerated phase, the leukemic ecosystem becomes more unstable, with increasing progenitor expansion, altered differentiation trajectories, epigenetic priming, and cooperating genetic lesions. Single-cell multi-omics is particularly valuable at this stage because it can separate clonal evolution from reversible transcriptional adaptation and can identify which progenitor states are becoming dominant before overt blast transformation[25,26].
In BC, CML biology becomes closer to acute leukemia, with profound differentiation blockade, expansion of immature blast-like states, increased chromatin accessibility at transcription factor programs such as MYC, TCF7L2, and GATA2, and stronger remodeling of the immune and stromal microenvironment[27,40]. Spatially resolved profiling further suggests that progression involves not only leukemic cell-intrinsic transformation but also vascular disorganization, immune exhaustion, and disruption of normal hematopoietic niches[40]. This phase-based view has direct clinical relevance: chronic-phase samples support risk stratification and early response prediction, accelerated-phase samples may reveal transition states suitable for early intervention, and blast-crisis samples help define combined therapeutic strategies targeting kinase signaling, epigenetic regulation, immune evasion, and supportive niches.
TRANSLATIONAL APPLICATIONS OF SINGLE-CELL SEQUENCING IN CML PRECISION DIAGNOSIS AND TREATMENT
Molecular heterogeneity and LSC subtyping of CML from a single-cell perspective
For a long time, enrichment strategies based on surface markers such as CD34 + CD38- have been used to initially extract LSCs. However, because normal HSCs also possess similar phenotypes, traditional bulk sequencing analysis is often confounded by a large amount of interference signals from normal cells[41,42]. This mixture of signals makes it difficult for researchers to identify rare resistant core clones that account for only a tiny proportion. Using single-cell transcriptomics combined with BCR::ABL1 targeted detection technology (BCR::ABL1 tSS2), Giustacchini et al. achieved the matching of fusion gene status and whole-transcriptome profiles at the single-cell level[43]. This study provided evidence that CML-LSCs are not a homogeneous population but exhibit substantial subpopulation differentiation and functional bias.
The study found that LSCs can be deconstructed into two core functional subpopulations: Group B, which features active proliferation, vigorous metabolism, and relative sensitivity to TKI killing; and Group A, which is highly quiescent with low transcriptional activity and enrichment of TGF-β and TNF-α inflammatory pathway characteristics[43]. Further functional genomics analysis showed that Group A exhibits increased survival resilience under TKI drug pressure by downregulating oxidative phosphorylation-related genes and upregulating pro-survival factors. The existence of Group A may contribute to the persistence of minimal residual disease (MRD) after treatment. Instead of being fully cleared under TKI pressure, these cells may show selective enrichment, constituting a potential seed bank for recurrence[43].
Further molecular analysis revealed that the Lin-CD34 + CD38- population can be subdivided into seven subpopulations with distinct gene expression patterns[44]. By dynamically tracking these subpopulations, researchers reported that the proportion of the Primitive Quiescent Cluster increased during TKI treatment. The molecular profile of this cluster showed inhibition of apoptotic pathways and activation of survival signals, particularly the synergistic effect of Wnt and Hedgehog pathways in maintaining stemness. To translate these single-cell findings into potentially translatable marker-based approaches, researchers identified a specific immunophenotypic combination, Lin-CD34 + CD38-CD45RA-cKIT-CD26+, through large-scale antibody screening as a candidate marker for identifying and targeting the resistant stem cell reservoir[44].
Additionally, using multi-level network analysis to integrate single-cell data, the study found molecular disturbances even in BCR::ABL1-negative residual stem cells. Under the long-term pressure of malignant clones, these seemingly "healthy" cells showed shifts in DNA damage repair pathways and metabolic homeostasis, suggesting that malignant clones may influence the broader bone marrow niche. Molecular features resolved by single-cell analysis—such as the role of S100A10 in promoting cell migration, the contribution of FCER1A to immune escape, and the involvement of RalA GTPase in maintaining LSC intracellular homeostasis—are more highly expressed in poor responders[45-48]. These findings provide possible mechanisms for understanding resistance and offer a molecular guide for exploring combined therapeutic targets beyond novel kinase inhibitors[47,48]. Notably, the FBXO3-DUSP9 axis has been identified via single-cell resolution as a potential mediator of LSC maintenance and TKI resistance[20].
Single-cell multidimensional analysis of the tumor microenvironment in CML
The pathogenesis of CML has evolved from a simple oncogene-driven model to a systemic perspective of "leukemia stem cell-tumor microenvironment" bidirectional regulation.
Inflammatory signal characteristics and prognostic value of the bone marrow microenvironment
Single-cell studies suggest that the bone marrow microenvironment of CML patients exhibits an inflammatory signal enrichment feature. Studies have found that inflammatory pathways such as IL-6, TGF-β, and TNF-α are upregulated in LSCs and their adjacent stromal cells[48,49]. The intensity of this inflammatory state may be negatively associated with the clinical efficacy of TKIs. At the diagnosis stage, high-intensity inflammatory features identified through single-cell sequencing, including high inflammatory signatures (e.g., TNF-α, NF-κB), may predict poorer molecular responses and shorter progression-free survival[16,17].
Regulatory mechanisms of inflammatory factors on LSC quiescence and differentiation fate
In-depth molecular mechanism analysis suggests that abnormally activated inflammatory signals may interfere with the cell cycle homeostasis and differentiation fate decisions of LSCs. Single-cell transcriptome trajectory analysis suggests that TGF-β can induce CML stem cells into a quiescent state by activating the SMAD signaling pathway and inhibiting the transcription of cell cycle-related genes[48]. Meanwhile, IL-6 may participate in paracrine and autocrine feedback loops. IL-6 secreted by myeloid leukemia cells acts on multipotent progenitor cells, inhibiting the normal B-lymphocyte differentiation pathway and reprogramming them toward myeloid differentiation via the activation of the transcription factor PU.1[49]. Additional niche-associated signaling programs may further modulate this process, but pathway-level extrapolation from non-CML models should be interpreted cautiously.
Impairment and recovery of immune surveillance functions
The abnormal inflammatory microenvironment of CML may also weaken host innate immune surveillance. Studies have found that this niche can inhibit the maturation process of natural killer (NK) cells and reduce their recognition and killing activity by downregulating the expression of surface activation receptors such as NKG2D[47,50,51]. However, successful TKI treatment is accompanied by remodeling of overall immune function. scRNA-seq suggests that functional recovery of CD56bright NK cells may support successful long-term drug discontinuation (TFR)[52,53]. In TFR patients, NK cells exhibit activation characteristics and express immune effector molecules such as CX3CR1, GZMB, and IFNG[52,53].
Application of single-cell sequencing in TKI resistance and clonal evolution research
Selective enrichment and kinetic patterns of resistant clones
The emergence of resistance often stems from the selection of resistant subclones already present at diagnosis. Single-cell kinetic analysis shows that TKIs preferentially reduce proliferative clones like Group B but have limited effect on Group A subpopulations. As treatment duration extends, the proportion of these quiescent BCR::ABL1 + LSCs may increase[43]. This process is reinforced by the epigenetic layer; under TKI pressure, the chromatin accessibility of stemness-related genes (such as Wnt/β-catenin pathway members and LEF1) increases in some studies[39,54,55]. Longitudinal single-cell tracking has further suggested that this progression may involve a continuous drift of transcriptional microstates[25]. These findings suggest therapeutic resistance potential from an epigenetic perspective[43,39].
Signaling pathway dysregulation and metabolic reprogramming promoting resistance
Resistant subpopulations exhibit unique transcriptomic features and metabolic patterns. Beyond the continuous activation of inflammatory pathways, the regulatory axis involving RalA may play an important role in resistant LSCs by improving DNA damage repair efficiency[36,46]. Furthermore, single-cell metabolic profiles reveal that resistant clones may shift from reliance on glycolysis to oxidative phosphorylation pathways and upregulate heat shock proteins (such as HSPB1) and autophagy-related genes to counter TKI-induced oxidative stress[43,51].
Dynamic changes in resistance-related immune markers
TKI treatment also alters the phenotypic evolution of LSC surface immune markers. Single-cell sequencing combined with flow cytometry validation found that residual LSCs after treatment often lose CD25 expression while continuing to maintain high CD26 levels[44]. This observation does not support CD25 as a universal eradication target for post-treatment residual LSCs; rather, CD25 should be interpreted as a state-dependent marker that may help define specific diagnostic or early-treatment subpopulations, whereas CD26, IL1RAP, CD93, and context-specific immune checkpoint ligands are more relevant for tracking persistent or resistant compartments[47,56]. Single-cell resolution has also identified rare resistant clones carrying NPM1 fusions that may contribute to drug resistance in specific clinical contexts[26,27]. These dynamically changing markers provide candidate targets for developing therapies against resistant LSCs, but their use requires careful attention to treatment timing, disease phase, and validation by orthogonal flow cytometry or proteomic assays.
Individualized treatment strategies informed by single-cell profiling
The clinical value of single-cell sequencing in CML is best understood as a stratification and decision-support framework rather than an immediate replacement for established molecular monitoring. At diagnosis, single-cell maps may help identify patients with an increased proportion of primitive quiescent BCR::ABL1-positive LSCs, inflammatory progenitor states, or immune-suppressive niches, thereby indicating a need for closer molecular monitoring, earlier mutation assessment, or consideration of combination strategies beyond TKI monotherapy[16,17,39]. During therapy, repeated or targeted single-cell profiling can distinguish pharmacologically suppressed proliferative clones from persisting quiescent LSC reservoirs, and can clarify whether suboptimal response reflects kinase-domain mutation, inflammatory niche protection, metabolic rewiring, or immune escape[20,21,43,44].
In practical terms, individualized strategies derived from single-cell evidence may include selecting patients for intensified monitoring when high-risk progenitor programs are present, prioritizing CD26- or IL1RAP-directed experimental approaches when residual stem-cell compartments are enriched, combining kinase inhibition with agents targeting inflammatory, apoptotic, metabolic, or epigenetic vulnerabilities, and using NK-cell or T-cell immune recovery signatures to support cautious treatment-free remission assessment[47,52,53,56]. These strategies remain translational rather than fully standardized, because most available studies are exploratory and require prospective validation, harmonized sample processing, and clinically interpretable reporting. The interplay among intrinsic LSC heterogeneity, niche regulation, and state-dependent markers is summarized in Figure 2.
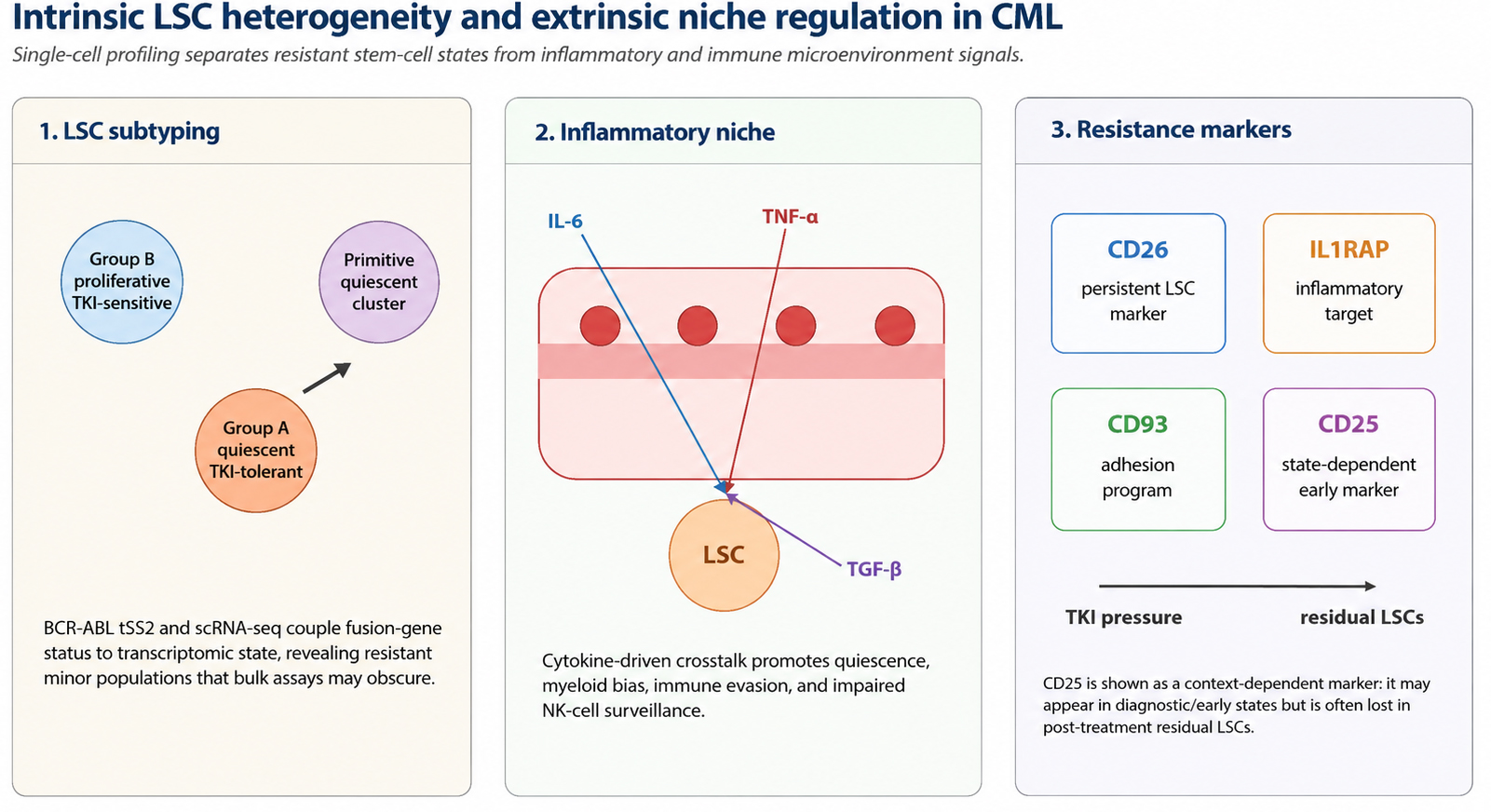
Figure 2. Interplay between intrinsic LSC heterogeneity and extrinsic microenvironmental regulation in CML. This illustration provides a panoramic view of CML precision management, spanning cellular subpopulation dynamics, inflammatory niche remodeling, and clinically relevant marker interpretation. Left panel: the Lin-CD34 + CD38- hematopoietic stem/progenitor cell compartment is resolved into functionally distinct sub-clusters via single-cell transcriptomics, including proliferative TKI-sensitive cells, quiescent TKI-tolerant cells, and primitive quiescent clusters. Middle panel: the leukemic bone marrow microenvironment is characterized by IL-6, TNF-alpha, and TGF-beta-mediated crosstalk that facilitates immune evasion and sustains the leukemic seed bank. Right panel: clinically relevant resistance-associated markers include CD26, IL1RAP, CD93, and state-dependent CD25 expression. CD25 is shown as an early or context-specific marker rather than a stable marker for post-treatment residual LSC eradication, because residual LSCs may lose CD25 while retaining CD26. This figure was redrawn by the authors using Python-based vector drawing and Microsoft Office-compatible elements; no third-party copyrighted materials were reused. LSC: Leukemia stem cell; TKI: tyrosine kinase inhibitor; scRNA-seq: single-cell RNA sequencing; IL-6: interleukin-6; TNF-alpha: tumor necrosis factor-alpha; TGF-beta: transforming growth factor-beta; IL1RAP: interleukin-1 receptor accessory protein.
FRONTIER INTEGRATION AND TECHNICAL SYNERGY OF SINGLE-CELL MULTI-OMICS
As single-cell transcriptomics research matures, the precision diagnosis and treatment of CML are rapidly evolving toward the deep integration of multi-omics and multi-modalities. This evolution is intended to construct a three-dimensional understanding of the entire life cycle of CML across multiple levels—including the source of gene regulation, epigenetic switches, protein functional execution, and physical interactions in spatial locations—thereby resolving resistance paradoxes that single-omics cannot explain.
Reconstruction of single-cell chromatin accessibility and epigenetic landscapes
As a cutting-edge single-cell epigenetic technique, single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq) enables high-resolution profiling of chromatin accessibility at the single-cell level, revealing epigenetic heterogeneity and regulatory networks underlying LSC maintenance and disease progression.
scATAC-seq has demonstrated useful advantages in parsing the maintenance mechanisms of LSCs by revealing which genomic regions are in a pre-activated state[50,57]. Research indicates that transcriptional dysregulation in LSCs is often pre-controlled by changes in chromatin openness. During the progression of CML toward the BC, scATAC-seq has observed enhanced accessibility at the binding sites of transcription factors such as MYC, TCF7L2, and GATA2[39,46,58].
By integrating scRNA-seq and scATAC-seq data, researchers can nominate enhancer regions and transcription-factor programs that may contribute to maintenance of the LSC quiescent state[39,57]. These integrated analyses should be interpreted as hypothesis-generating tools for pathway prioritization rather than definitive proof of a single inflammatory or epigenetic axis.
Frontier atlas of single-cell proteomics and multi-omics integration
In the molecular regulatory network of CML, transcript-level abundance does not always linearly predict the final functional product. Single-cell proteomics therefore occupies an important complementary position.
Currently, single-cell proteo-transcriptomics (such as CITE-seq) allows for the parallel analysis of cell surface protein abundance and mRNA levels. As demonstrated in CML single-cell studies, this proteogenomic profiling can define stem and progenitor cell sub-clusters more precisely and may bridge the gap between sequencing-based discovery and clinically familiar immunophenotyping[16,39]. Furthermore, studies on early cytoreductive therapies, such as hydroxyurea, show that single-cell profiles can sensitively capture changes in the stress states of cells under pharmacological intervention[18].
In the dimension of signaling pathway monitoring, Cytometry by Time-of-Flight (CyTOF) enables the simultaneous monitoring of over 40 surface markers and kinase phosphorylation states[59,60]. Recent integrated proteogenomic analyses suggest that the PI3K/AKT/mTOR signaling pathway is a common cross-disease therapeutic vulnerability, where protein phosphorylation levels may be associated with leukemic transformation risk and may help stratify patients in broader myeloid contexts[9]. Regarding resistance mechanisms, dynamic single-cell tracking has revealed that resistance to novel drugs like Selinexor is closely associated with inhibition of the intracellular ferroptosis pathway[21].
In the genomic dimension, the introduction of joint analysis of mutations and transcriptomes (scWGS or long-read sequencing) is redefining clonal evolution research[51,61]. This allows researchers to identify "preferred transcriptional states" within complex clonal backgrounds—how specific mutant clones (such as those with NPM1 fusions or specific BCR::ABL1 mutations) gain expansion or resistance advantages[26,27].
Finally, single-cell technology is moving toward spatial multi-omics. Through 3D imaging and spatial transcriptomics, including CODEX-based approaches, CML-focused studies suggest that even BCR::ABL1-negative bystander cells, such as macrophages, can undergo transcriptional and functional reprogramming under leukemic environmental pressure, thereby contributing to immune evasion and niche support[40,62]. Outside the CML-specific evidence base, comprehensive molecular profiling frameworks such as the MOTION study offer a general example of how multi-omics data may be organized into interpretable molecular characterization pipelines; this evidence should be viewed as broader translational context rather than direct CML-specific support for treatment selection[63]. Together, these observations encourage a systematic ecological interpretation of CML in which leukemic clones, immune cells, stromal elements, vasculature, and spatial niches are analyzed together. The technological synergy among scATAC-seq, CODEX, CITE-seq, CyTOF, spatial omics, and computational modeling is summarized in Figure 3.
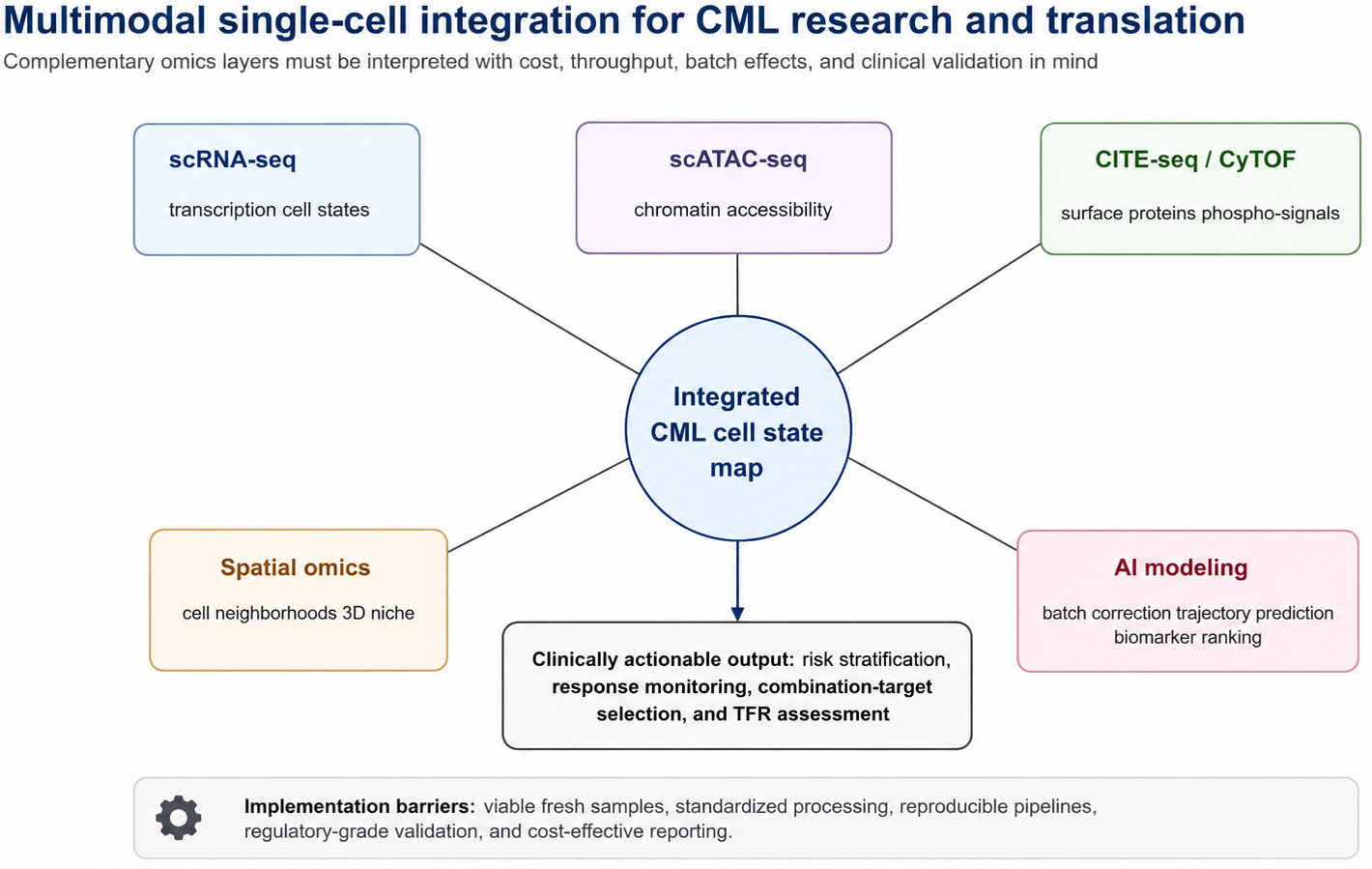
Figure 3. Technological synergy and multimodal data integration in CML research. A comprehensive framework demonstrating the convergence of diverse single-cell and multimodal technologies to resolve the biological complexity of CML. scRNA-seq defines transcriptional cell states; scATAC-seq maps chromatin accessibility and epigenetic regulatory programs; CITE-seq and CyTOF facilitate parallel profiling of surface protein abundance and intracellular signaling pathways; spatial omics, including CODEX-based approaches where available, helps characterize cell neighborhoods and three-dimensional bone marrow niche organization; and AI-assisted computational modeling supports batch correction, trajectory prediction, and biomarker prioritization. This integrated approach enables a holistic understanding of CML across multiple biological scales, from gene regulation and epigenetic states to protein execution, spatial organization, and clinically interpretable decision support. This figure was redrawn by the authors using Python-based vector drawing and Microsoft Office-compatible elements; no third-party copyrighted materials were reused. scRNA-seq: Single-cell RNA sequencing; scATAC-seq: single-cell assay for transposase-accessible chromatin using sequencing; CODEX: co-detection by indexing; CITE-seq: cellular indexing of transcriptomes and epitopes by sequencing; CyTOF: cytometry by time-of-flight; 3D: three-dimensional.
Computational interpretation, artificial intelligence, and translational barriers
Artificial intelligence and machine learning can be integrated into the body of single-cell CML research rather than appearing only as a future aspiration. In current analytical workflows, machine learning is used for quality control, cell annotation, batch correction, trajectory inference, regulatory-network reconstruction, feature prioritization, and response prediction. Deep generative frameworks such as scVI provide probabilistic integration of single-cell datasets while accounting for dropout, library size, and batch effects[64]. Foundation models such as scGPT further illustrate how large-scale pretrained representations may support cell-type annotation, perturbation modeling, and cross-dataset transfer learning, although their reliability in rare hematologic malignant states still requires benchmarking and interpretability assessment[65]. In CML specifically, AI-enabled single-cell analysis pipelines have begun to combine gene regulatory networks, trajectory inference, and gene signature discovery to analyze NK-cell programs associated with treatment-free remission and relapse after TKI discontinuation[66].
Despite this promise, clinical implementation remains limited by several barriers. Single-cell sequencing often requires viable fresh or carefully preserved samples, rapid processing, sufficient cell numbers, and harmonized protocols to avoid artifactual shifts in cell-state composition. Batch effects, doublets, dropout, dissociation-induced stress signatures, antibody-panel variability, and differences in sequencing depth can all distort biological interpretation. Data analysis also remains complex, requiring transparent pipelines, reference atlases, validated thresholds, and clinically interpretable reports rather than exploratory clusters alone. Costs, turnaround time, reimbursement, regulatory review, laboratory accreditation, and data privacy requirements further restrict routine use. Therefore, single-cell technologies currently function primarily as discovery, stratification, and translational research tools; their integration into routine CML care should proceed through prospective validation, standardized reporting, and comparison against established BCR::ABL1 quantitative PCR, mutation testing, flow cytometry, and measurable residual disease assays.
CONCLUSION AND PROSPECTS
Single-cell sequencing technology has brought important advances to CML research by moving interpretation from averaged molecular signals toward cell-state-resolved biology. The evidence reviewed here indicates that therapeutic response and relapse risk are shaped by quiescent LSC reservoirs, progenitor differentiation bias, immune remodeling, metabolic adaptation, and spatial niche protection rather than by BCR::ABL1 burden alone. The principal contribution of this review is to integrate these observations into a cautious translational framework in which single-cell profiles complement, but do not replace, established molecular monitoring.
For future clinical use, the most immediate value is likely to lie in refining risk stratification, interpreting suboptimal TKI response, identifying residual disease programs that may require combination strategies, and supporting carefully selected treatment-free remission assessment. These applications should be pursued with standardized sampling, reproducible computational workflows, predefined reporting thresholds, and cross-platform validation. AI-assisted analysis may improve annotation and biomarker prioritization, but it should remain transparent and externally validated before affecting patient management.
Overall, single-cell technology offers a promising but still developing route toward more individualized CML management. Its clinical impact will depend on prospective evidence, cost and turnaround control, regulatory-grade quality assurance, and clear integration with BCR::ABL1 quantitative PCR, mutation testing, flow cytometry, and measurable residual disease assays. In this sense, the field should move from descriptive atlases toward clinically testable decision-support models while avoiding premature claims of routine actionability[67-70].
DECLARATIONS
Acknowledgment
The authors thank Prof. Junjiang Fu and Prof. Tao He for their valuable comments, guidance, and support during the revision of this manuscript.
Author contribution
Conceptualization, literature synthesis, supervision, visualization, and writing - original draft: Li X
Literature search, evidence organization, and writing support: Chen R
Conceptual guidance and supervision: Huang Y
Literature search: Xiao X
Resources, visualization support, and writing – review & editing: Wu L
Conceptualization, conceptual guidance, supervision, and writing - review & editing: Zhu X
Conceptualization, project administration, supervision, critical interpretation, visualization, and writing - review & editing: Liu J
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2025 update on diagnosis, therapy, and monitoring. Am J Hematol. 2024;99:2191-212.
2. Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and giemsa staining. Nature. 1973;243:290-3.
3. Faderl S, Talpaz M, Estrov Z, O'brien S, Kurzrock R, Kantarjian HM. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164-72.
4. Mandanas R, Leibowitz D, Gharehbaghi K, et al. Role of p21 RAS in p210 bcr-abl transformation of murine myeloid cells. Blood. 1993;82:1838-47.
5. Minciacchi VR, Kumar R, Krause DS. Chronic myeloid leukemia: a model disease of the past, present and future. Cells. 2021;10:117.
6. Raitano AB, Halpern JR, Hambuch TM, Sawyers CL. The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci USA. 1995;92:11746-50.
7. Stein SJ, Baldwin AS. NF-κB suppresses ROS levels in BCR-ABL+ cells to prevent activation of JNK and cell death. Oncogene. 2011;30:4557-66.
8. Tam WF, Gu T, Chen J, et al. Id1 is a common downstream target of oncogenic tyrosine kinases in leukemic cells. Blood. 2008;112:1981-92.
9. He F, Lin S, Gao B, et al. A proteogenomic gene signature defines prognostic subgroups highlighting PI3K/AKT/mTOR signaling pathway as a therapeutic vulnerability in myeloid malignancies. Cell Commun Signal. 2025;24:61.
10. Mohammadhosseini M, Enright T, Duvall A, et al. Targeting the CD74 signaling axis suppresses inflammation and rescues defective hematopoiesis in RUNX1-familial platelet disorder. Sci Transl Med. 2025;17:eadn9832.
11. Huang X, Cortes J, Kantarjian H. Estimations of the increasing prevalence and plateau prevalence of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Cancer. 2012;118:3123-7.
12. Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129-41.
13. Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399-401.
14. Druker BJ, Lydon NB. Lessons learned from the development of an Abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Investig. 2000;105:3-7.
15. Jabbour E, Sasaki K, Haddad FG, et al. Low‐dose dasatinib 50 mg/day versus standard‐dose dasatinib 100 mg/day as frontline therapy in chronic myeloid leukemia in chronic phase: a propensity score analysis. Am J Hematol. 2022;97:1413-8.
16. Krishnan V, Schmidt F, Nawaz Z, et al. A single-cell atlas identifies pretreatment features of primary imatinib resistance in chronic myeloid leukemia. Blood. 2023;141:2738-55.
17. Zhang W, Yang B, Weng L, et al. Single cell sequencing reveals cell populations that predict primary resistance to imatinib in chronic myeloid leukemia. Aging. 2020;12:25337-55.
18. Komic H, Nilsson MS, Wennström L, et al. Single-cell proteo-transcriptomic profiling reveals altered characteristics of stem and progenitor cells in patients receiving cytoreductive hydroxyurea in early-phase chronic myeloid leukemia. Haematologica. 2025;110:117-28.
19. Stengel A, Hörst K, Kühn C, et al. Potential factors underlying the progression of RUNX1-mutated MDS to AML. Cancer Genet. 2025;294-5:181-3.
20. Li X, Zuo S, Zhang Y, et al. FBXO3-mediated DUSP9 ubiquitination promotes leukemia stem cell maintenance and tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Cell Rep Med. 2026;7:102686.
21. Cui Y, Li Y, Ji J, et al. Dynamic single-Cell RNA-seq reveals mechanism of selinexor-resistance in chronic myeloid leukemia. Int Immunopharmacol. 2024;134:112212.
22. Liu H, Li Y, Karsidag M, Tu T, Wang P. Technical and biological biases in bulk transcriptomic data mining for cancer research. J Cancer. 2025;16:34-43.
23. Munje C, Copland M. Exploring stem cell heterogeneity in chronic myeloid leukemia. Trends Cancer. 2018;4:167-9.
24. Zheng Y, Yang X. Spatial RNA sequencing methods show high resolution of single cell in cancer metastasis and the formation of tumor microenvironment. Biosci Rep. 2023;43:BSR20221680.
25. Frankhouser DE, Zhao D, Fu YH, et al. Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia. Syst Biol. 2025:2025.05.14.653262.
26. Cho J, Cao J, Hemberg M. Joint analysis of mutational and transcriptional landscapes in human cancer reveals key perturbations during cancer evolution. Genome Biol. 2024;25:65.
27. Zhu L, Yi K, Zhou J, et al. Concurrent NPM1::CCDC28A and BCR::ABL1 fusions in extramedullary blast crisis of chronic myeloid leukemia: A case report and literature review. Ann Hematol. 2025;104:6045-51.
28. Dawson A, Zarou MM, Prasad B, et al. Leukaemia exposure alters the transcriptional profile and function of BCR::ABL1 negative macrophages in the bone marrow niche. Nat Commun. 2024;15:1090.
29. Zhuo C, Dong X, Zhao X, et al. Single-cell sequencing reveals the expansion and diversity of T cell subsets in the bone marrow microenvironment of chronic myeloid leukemia. Genes Dis. 2025;12:101626.
30. Svensson V, Vento-Tormo R, Teichmann SA. Exponential scaling of single-cell RNA-seq in the past decade. Nat Protoc. 2018;13:599-604.
31. Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformaticspipelines. Exp Mol Med. 2018;50:1-14.
32. Shaffer SM, Dunagin MC, Torborg SR, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431-5.
33. Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377-82.
34. Salmen F, De Jonghe J, Kaminski TS, et al. High-throughput total RNA sequencing in single cells using VASA-seq. Nat Biotechnol. 2022;40:1780-93.
35. Hagemann-Jensen M, Ziegenhain C, Chen P, et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat Biotechnol. 2020;38:708-14.
36. Gierahn TM, Wadsworth MH, Hughes TK, et al. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods. 2017;14:395-8.
37. Fan HC, Fu GK, Fodor SPA. Combinatorial labeling of single cells for gene expression cytometry. Science. 2015;347:1258367.
38. Wang X, He Y, Zhang Q, Ren X, Zhang Z. Direct comparative analyses of 10X genomics chromium and smart-Seq2. Genom Proteom Bioinform. 2021;19:253-66.
39. Warfvinge R, Geironson Ulfsson L, Dhapola P, et al. Single-cell multiomics analysis of chronic myeloid leukemia links cellular heterogeneity to therapy response. eLife. 2024;12:RP92074.
40. Li L, Rottmann I, Saeed BR, et al. High-dimensional spatiotemporal single-cell atlas and 3D imaging of the bone marrow microenvironment during CML progression. Blood. 2026;147:2648-65.
41. Mustjoki S, Richter J, Barbany G, et al. Impact of malignant stem cell burden on therapy outcome in newly diagnosed chronic myeloid leukemia patients. Leukemia. 2013;27:1520-6.
42. Sloma I, Beer PA, Saw KM, et al. Genotypic and functional diversity of phenotypically defined primitive hematopoietic cells in patients with chronic myeloid leukemia. Exp Hematol. 2013;41:837-47.
43. Giustacchini A, Thongjuea S, Barkas N, et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat Med. 2017;23:692-702.
44. Warfvinge R, Geironson L, Sommarin MNE, et al. Single-cell molecular analysis defines therapy response and immunophenotype of stem cell subpopulations in CML. Blood. 2017;129:2384-94.
45. Ma J, Pettit N, Talburt J, Wang S, Weissman SM, Yang MQ. Integrating single-cell transcriptome and network analysis to characterize the therapeutic response of chronic myeloid leukemia. Int J Mol Sci. 2022;23:14335.
46. Yin Z, Su R, Ge L, et al. Single-cell resolution reveals RalA GTPase expanding hematopoietic stem cells and facilitating of BCR-ABL1-driven leukemogenesis in a CRISPR/Cas9 gene editing mouse model. Int J Biol Sci. 2023;19:1211-27.
47. Patterson SD, Copland M. The bone marrow immune microenvironment in CML: treatment responses, treatment-free remission, and therapeutic vulnerabilities. Curr Hematol Malig Rep. 2023;18:19-32.
48. Nievergall E, Reynolds J, Kok CH, et al. TGF-α and IL-6 plasma levels selectively identify CML patients who fail to achieve an early molecular response or progress in the first year of therapy. Leukemia. 2016;30:1263-72.
49. Reynaud D, Pietras E, Barry-Holson K, et al. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell. 2011;20:661-73.
50. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213-8.
51. Chimge N, Chen M, Nguyen C, et al. A deeply quiescent subset of CML LSC depend on FAO yet avoid deleterious ROS by suppressing mitochondrial complex I. Curr Mol Pharmacol. 2023;17:e060923220758.
52. Yu G, Lu W, Chen X, et al. Single-cell RNA sequencing to explore composition of peripheral blood NK cells in patients with chronic myeloid leukemia in treatment-free remission. Leukemia Lymphoma. 2022;63:2604-15.
53. Imeri J, Desterke C, Marcoux P, et al. Case report: long-term voluntary Tyrosine Kinase Inhibitor (TKI) discontinuation in chronic myeloid leukemia (CML): molecular evidence of an immune surveillance. Front Oncol. 2023;13:1117781.
54. Bertacchini J, Ketabchi N, Mediani L, Capitani S, Marmiroli S, Saki N. Inhibition of Ras-mediated signaling pathways in CML stem cells. Cell Oncol. 2015;38:407-18.
55. Zhou J, Zhao Y, Leng J, et al. DNA methylation-mediated differential expression of DLX4 isoforms has opposing roles in leukemogenesis. Cell Mol Biol Lett. 2022;27:59.
56. Holyoake TL, Vetrie D. The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood. 2017;129:1595-606.
57. Perry JM, Tao F, Roy A, et al. Overcoming Wnt-β-catenin dependent anticancer therapy resistance in leukaemia stem cells. Nat Cell Biol. 2020;22:689-700.
58. Desterke C, Hugues P, Hwang JW, Bennaceur-Griscelli A, Turhan AG. Embryonic program activated during blast crisis of chronic myelogenous leukemia (CML) implicates a TCF7L2 and MYC cooperative chromatin binding. Int J Mol Sci. 2020;21:4057.
59. Veenstra J, Dimitrion P, Yao Y, Zhou L, Ozog D, Mi Q. Research techniques made simple: use of imaging mass cytometry for dermatological research and clinical applications. J Investig Dermatol. 2021;141:705-12.e1.
60. Cheung TK, Lee C, Bayer FP, Mccoy A, Kuster B, Rose CM. Defining the carrier proteome limit for single-cell proteomics. Nat Methods. 2020;18:76-83.
61. Coorens THH, Oh JW, Choi YA, et al. The somatic mosaicism across human tissues network. Nature. 2025;643:47-59.
62. Rao A, Barkley D, França GS, Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature. 2021;596:211-20.
63. Kuznetsova OA, Ivanov MV, Lebedeva AA, et al. Comprehensive molecular profiling in MOTION study. Cancer Genet. 2025;296-7:45-52.
64. Lopez R, Regier J, Cole MB, Jordan MI, Yosef N. Deep generative modeling for single-cell transcriptomics. Nat Methods. 2018;15:1053-8.
65. Cui H, Wang C, Maan H, et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat Methods. 2024;21:1470-80.
66. Borra S, Yan D, Welner RS, Yue Z. An AI-enabled single-cell transcriptomic analysis pipeline for gene signature discovery in natural killer cells linked to remission outcomes in chronic myeloid leukemia. Biology. 2026;15:588.
67. Huuhtanen J, Adnan-Awad S, Theodoropoulos J, et al. Single-cell analysis of immune recognition in chronic myeloid leukemia patients following tyrosine kinase inhibitor discontinuation. Leukemia. 2023;38:109-25.
68. Lei Y, Tang R, Xu J, et al. Applications of single-cell sequencing in cancer research: progress and perspectives. J Hematol Oncol. 2021;14:91.
69. Hu Y, An Q, Sheu K, Trejo B, Fan S, Guo Y. Single cell multi-omics technology: methodology and application. Front Cell Dev Biol. 2018;6:28.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].