



Advances in mRNA-LNP lung-targeted delivery strategies
 0
0 
Abstract
The lung, a vital organ for homeostasis, is vulnerable to various diseases that challenge healthcare systems due to limited treatment options. Fortunately, mRNA-based gene therapy offers a promising solution, demonstrating high efficiency and safety across applications in vaccines, protein replacement therapy, and cancer treatment. However, naked mRNA faces challenges like degradation, poor cell penetration, and immunogenicity. The lung’s complex structure further complicates mRNA delivery. In this way, lipid nanoparticles (LNPs) have emerged as an effective solution, demonstrated by their success in COVID-19 mRNA vaccines through superior encapsulation and biocompatibility. Extensive studies focus on developing LNP-based pulmonary mRNA delivery systems for treating viral infections and lung diseases.This review analyzes the current state and developments in mRNA-LNP applications for pulmonary diseases and LNP-based strategies for lung-targeted mRNA delivery. We explore the optimization and development of LNP platforms across four administration routes: nebulized inhalation, intratracheal administration, nasal administration, and systemic administration. Our goal is to provide researchers with a comprehensive reference covering both fundamental principles and cutting-edge developments in pulmonary mRNA-LNP delivery systems.

Keywords
INTRODUCTION
The lung is a cornerstone organ in maintaining human physiological balance and homeostasis[1]. Meanwhile, the lung is a primary gateway wherein harmful environmental substances might enter the human body, which makes it particularly vulnerable to various pathological conditions[2]. Many diseases specifically target the lung, such as viral/bacterial infections[3], acute lung injury[4], chronic obstructive pulmonary disease (COPD)[5], cystic fibrosis (CF)[6], and lung cancer[7]. These diseases cause pulmonary dysfunction, severely impacting patients' quality of life and threatening human health. Nowadays, lung diseases represent a substantial burden on healthcare systems worldwide - COPD alone caused 4 million deaths globally in 2019[8]. Moreover, contemporary clinical medicine faces significant challenges in providing effective therapeutic solutions, with many current treatment options failing to deliver satisfactory outcomes for these devastating conditions.
Fortunately, with the rapid advancement of biomedical technology, mRNA-based gene therapy has emerged as an exceptionally promising approach for addressing various pulmonary[9]. mRNA therapy facilitates the temporary expression of specific proteins within cells, representing a significant breakthrough in gene therapy. On the one hand, mRNA therapy substantially enhances the expression efficiency of protein. More importantly, it eliminates concerns about genomic contamination, which is the most critical safety consideration in gene therapy applications. The versatility of mRNA therapy has led to remarkable achievements across multiple fields, including vaccine development for infectious diseases, protein replacement strategies for genetic disorders, and novel cancer treatments.
Particularly, mRNA therapy demonstrates transformative potential in vaccine development[10]. Compared to other technologies, mRNA offers multiple advantages, including the ability to encode multiple protein targets simultaneously, standardized production protocols, and streamlined purification processes. These advantages have dramatically shortened vaccine development timelines, enabling rapid responses to emerging health threats. The COVID-19 pandemic demonstrated this technology’s profound impact, as rapidly deployed mRNA-based vaccines saved millions of lives worldwide.
However, naked mRNA is not only easily degraded in vivo and difficult to penetrate cell membranes, but also has strong immunogenicity, readily activating the body’s innate immune response and triggering inflammation[11,12]. These drawbacks severely limit its clinical application development. Therefore, developing effective mRNA delivery platforms is crucial. Currently, common mRNA delivery vectors include various biological derivatives (such as virus-like particles, bacterial outer membrane vesicles, and exosomes) and chemical derivatives (polymer and lipid-based nanoparticles). Among these, lipid nanoparticles (LNPs) have garnered attention due to their excellent encapsulation efficiency and biocompatibility. Given their success in COVID-19 mRNA vaccines, LNPs are considered the optimal platform for mRNA delivery.
Furthermore, the unique anatomical structure and physiological characteristics of the lungs present challenges for pulmonary mRNA delivery. In this way, LNPs serve as an excellent platform for targeting mRNA delivery to lung tissue[13]. Extensive research is dedicated to developing LNP-based pulmonary mRNA delivery systems to prevent viral infections and treat lung diseases.
In this review, we present a detailed analysis of the current state and ongoing developments in mRNA-LNP applications for pulmonary diseases, along with an extensive examination of innovative LNP-based strategies specifically designed for lung-targeted mRNA delivery. We focus on the methodological optimization and strategic development of LNP platforms, based on the unique requirements of different administration routes of lung-targeted mRNA delivery. To provide a thorough understanding of this field, we summarized four different administration routes: nebulized inhalation, intratracheal administration, nasal administration, and systemic administration. For each approach, we highlight the most recent research breakthroughs and technological innovations. We aim to provide researchers in this rapidly evolving field with a valuable reference that encompasses both fundamental principles and cutting-edge developments in pulmonary mRNA-LNP delivery systems.
INTRODUCTION TO LNP
LNPs are nanoscale lipid-based carriers that can encapsulate various types of nucleic acids, protecting them from degradation by the immune system while also facilitating cellular uptake. LNPs enter cells via endocytosis and subsequently release their nucleic acid cargo into the cytoplasm through the endosomal escape process. This mechanism enables therapeutic effects such as protein expression or gene silencing[14]. In recent years, RNA-based therapies utilizing LNPs have gained significant momentum. Notably, the U.S. Food and Drug Administration (FDA) approved Onpattro® in 2018, the first LNP-based gene therapy, for the treatment of transthyretin-mediated amyloidosis. Furthermore, during the COVID-19 pandemic, mRNA-LNP vaccines, such as mRNA-1273 and BNT162b2, garnered considerable attention and were successfully commercialized[15,16]. These applications establish LNPs’ central position in targeted delivery. In this section, we provide a comprehensive overview of LNPs, focusing on their main components, preparation methods, and advantages [Figure 1].

Figure 1. Introduction of mRNA-LNP: main components, preparation methods and advantages. LNP: Lipid nanoparticle.
Main components
LNPs are primarily composed of four key components: ionizable cationic lipids, cholesterol, phospholipids, and PEGylated lipids[17-19]. Each component plays a crucial role in ensuring efficient encapsulation, stability, and delivery of mRNA.
Ionizable cationic lipids are crucial for encapsulating mRNA and are the core component of LNPs. These lipids typically consist of a head group, a linker, and a hydrophobic tail. The head group, containing amine, guanidine, and heterocyclic groups, promotes mRNA encapsulation, stabilizes nanoparticles, interacts with cell membranes, and facilitates endosomal escape. The linker influences the biodegradability, cellular tolerance, and delivery efficiency. The hydrophobic tail affects the pKa and fluidity of the ionizable lipids, which in turn impacts LNP function[20]. In the neutral extracellular environment, ionizable cationic lipids are positively charged, allowing them to bind with the negatively charged mRNA, thereby improving encapsulation. Upon cellular uptake, the drop in pH from approximately 6.2 to 4.9-6.0 triggers the protonation of these lipids, facilitating the release of mRNA from the endosome[21].
Cholesterol, as a structural component, aids in particle formation and enhances the stability of the lipid bilayer and membrane fusion[22]. Optimizing the cholesterol structure can further improve the delivery efficiency of LNPs and promote endosomal escape. Jung et al.[23] developed an ionizable helper cholesterol analog, 3β[L-histidinamide-carbamoyl] cholesterol (Hchol) lipid, which significantly improved mRNA escape from endosomes and delivery efficiency.
Phospholipids, such as dioleoylphosphatidylcholine (DOPC) and dioleoylphosphatidylethanolamine (DOPE), are auxiliary components that support mRNA packaging and stabilize the LNP structure[24]. These phospholipids typically make up 10%-20% of the total lipid content[25].
PEGylated lipids improve the biocompatibility and stability of LNPs, enhancing their ability to deliver drugs and genes to target cells and tissues while preventing aggregation during transport[26,27]. Generally, longer polyethylene glycol (PEG) lengths reduce non-specific interactions with serum components, thereby extending the half-life of LNPs. Nevertheless, long PEG lipids inhibit the fusion between LNPs and endosomal membranes, reducing delivery efficiency. This contradiction is known as the “PEG dilemma” in LNP drug delivery[28]. Moreover, PEGylated lipids, despite their low immunogenicity, may induce the production of specific antibodies such as IgM, IgG, and IgE, leading to accelerated clearance of LNPs from the bloodstream. Additionally, when PEGylated lipids interact with certain proteins or nanoparticles, they can also cause hypersensitivity reactions such as complement activation-related pseudoallergy[29,30].
In summary, the four main components of LNPs - ionizable cationic lipids, cholesterol, phospholipids, and PEGylated lipids - work in concert to ensure the effective encapsulation, stability, intracellular delivery, and endosomal escape of mRNA. The precise ratio and structural design of these components are crucial for the performance of LNPs, determining their distribution in the body, cellular uptake, and, ultimately, the therapeutic effect. Therefore, optimizing these components is essential for developing efficient and safe mRNA delivery systems.
Preparation methods
The preparation of LNPs is based on the principle of self-assembly. First, ionizable cationic lipids become protonated in an acidic environment and carry a positive charge. This allows them to interact electrostatically with negatively charged mRNA, facilitating the encapsulation of the mRNA within the LNPs. Other lipids (including phospholipids, cholesterol, and PEGylated lipids) self-assemble on this basis to stabilize the formed structure. Following self-assembly, the mRNA-LNP’s PH solution is adjusted to neutral with a buffer exchange, which can remove the ethanol solvent at the same time. As the ionizable lipids become uncharged at physiological pH, the LNPs become more stable and less toxic[31,32]. To improve this process, Geng et al.[33] introduced a two-step tangential flow filtration (TFF) method. In the traditional single-step TFF method, ethanol is removed simultaneously with pH adjustment, which causes rapid changes in the ionization state of the cationic lipids and results in incomplete fusion, leading to a heterogeneous population of smaller particles and a higher proportion of empty LNPs. In contrast, the two-step TFF method first removes ethanol at a low pH, reducing electrostatic repulsion between particles and allowing controlled fusion to occur more gradually when the pH is subsequently increased. Therefore, mRNA-LNPs prepared by the two-step TFF method have larger particle sizes and lower empty LNP ratios, and their storage stability is optimized.
Several methods have been developed for preparing LNPs, including microfluidic technology, jet mixing, and solvent injection, often utilizing robotic liquid handling devices.
Microfluidic technology
Microfluidic technology involves the precise manipulation of liquids at the sub-millimeter scale using microscale devices. This technique allows for rapid sample processing with high precision[34]. Microfluidic devices have two inlets for the entry of lipid/ethanol solutions and RNA/buffer solutions. Lipids and mRNA are dissolved in ethanol and acidic aqueous phases, respectively, and the ethanol and aqueous phases are mixed in a volume ratio of 1:3 through the microfluidic device to complete the self-assembly process. Compared with traditional bulk methods, microfluidic technology can generate highly stable, uniform, and monodisperse particles, with reagent consumption reduced to picoliters and reaction times reduced to seconds[35]. However, scaling microfluidic technology from laboratory to industrial production may face technical and cost challenges. Shepherd et al.[36] proposed a scalable parallel microfluidic device that achieved more than 100 times the production rate compared to a single microfluidic channel, which, to some extent, promotes the possibility of industrial production using microfluidic technology.
Jet mixing method
Although microfluidic technology has been widely applied, scaling up to industrial production is challenging. As a result, the jet mixing method is more commonly used in large-scale LNP production. This method replaces microfluidic chips with mixers such as restricted impinging jets (CIJs), multi-inlet vortex mixers (MIVMs), or co-axial turbulent jet mixers, all operating at the micrometer scale. The turbulent conditions in these mixers facilitate rapid mixing and control LNP size and distribution[37]. Studies indicate that co-axial turbulent jet mixers produce smaller and more uniform LNPs than microfluidic devices. However, these mixers are an order of magnitude larger in volume, with issues like large footprint and high cost, limiting their research application[38].
Robotic liquid handling devices
Robotic liquid handling devices, such as TECAN EVO, are emerging as an efficient method for LNP preparation. In this approach, an acidic buffer solution containing RNA (pH 4.0) and a lipid mixture is mixed in a 96-well plate, allowing the LNPs to self-assemble. The robotic liquid handling devices inject and mix into the wells of the plate at specific speeds (for example, 500 µL/s) and loop times (for example, 10 loops, each loop 100 µL) to ensure effective mixing of lipids and RNA, thus forming LNPs[39]. One major advantage of robotic liquid handling devices is their ability to process up to 384 different LNP formulations simultaneously, while microfluidic technology typically prepares only one formulation at a time. This high throughput is particularly useful for accelerating the development and optimization of proprietary lipid formulations, as well as for mRNA LNP lead compound optimization and preclinical candidate drug selection[40].
Advantages
Building on the advances in LNP preparation methods, the advantages of LNPs as a carrier for mRNA delivery are increasingly evident. Currently, LNPs represent the most advanced mRNA delivery platform in clinical use, offering several significant benefits as a non-viral vector[25].
Firstly, LNPs exhibit a high safety profile. Unlike viral vectors, which may possess high cytotoxicity and immunogenicity, including the potential risk of oncogenesis, LNPs are generally considered safe. This makes them ideal for repeated dosing and long-term treatments[13,41]. Cationic lipids, a key component of LNPs, can be used to encapsulate mRNA, but permanently positively charged cationic lipids may be cytotoxic[42]. Therefore, researchers have developed functionally identical and relatively safe ionizable lipids[43]. As the composition of LNPs continues to improve, their safety profile has also become more robust.
Moreover, LNPs protect mRNA from degradation, thereby extending its circulation time in the body and enhancing both its stability and half-life, significantly improving the efficiency of mRNA delivery. In vitro transfection experiments using primary dendritic cells (DCs) have shown that LNPs can effectively transfect DCs with an efficiency increase of up to 450%, while naked mRNA fails to achieve successful transfection[44]. Encapsulating mRNA within LNP carriers thus markedly improves delivery efficiency[45].
Furthermore, LNPs can achieve targeted delivery of mRNA in vivo, including targeting specific organs and specific cell populations[46]. A variety of targeting strategies have been developed, including optimizing the prescription design of LNPs or modifying their surfaces differently, which can change their affinity for specific cell types in the body and achieve targeted delivery of mRNA[47].
Finally, LNPs are relatively simple to design and synthesize, and the production process is relatively direct, with a straightforward production process, facilitating industrial production and reducing costs[48]. Additionally, LNPs can encapsulate various mRNA molecules, enabling quick response to changes in pathogens[49]. Therefore, LNPs are an ideal platform for the rapid development of vaccines, enabling quick reactions to new epidemics.
In this review, we focus on the use of LNP in mRNA delivery. The effective delivery of mRNA-LNPs to lung cells requires overcoming several key cellular barriers, including interface barrier, intracellular trafficking barrier, and targeting barrier[50]. For effective drug delivery, nanocarriers must be able to enter the cell and escape from endosomes. Due to the negatively charged nature of the cell membrane, LNPs are engineered with ionizable lipids that facilitate endocytosis. However, once internalized, mRNA-LNPs face degradation within lysosomes, needing functional materials such as protonatable lipids to induce endosomal escape via the proton sponge effect[42]. Moreover, ligand-functionalized LNPs enable active targeting of lung-specific receptors, improving transfection efficiency and tissue specificity. These designs ensure effective targeting and release within lung cells by optimizing the design of the LNP, particularly by tuning the surface properties of the nanoparticles and decorating them with specific ligands. Ultimately, the cellular barrier can be overcome to reach and function[51].
LNPs have become pivotal in the delivery of nucleic acid-based therapeutics, leading to the approval of several groundbreaking drugs. In 2018, the U.S. FDA approved Patisiran (brand name Onpattro), the first LNP-formulated siRNA therapeutic developed by Alnylam Pharmaceuticals[52]. In 2020, two mRNA-based COVID-19 vaccines utilizing LNP technology were authorized: BNT162b2 (Comirnaty) by Pfizer/BioNTech and mRNA-1273 (Spikevax) by Moderna[53]. In 2024, Moderna’s mRNA-1345 (mRESVIA), a vaccine targeting respiratory syncytial virus (RSV), was also approved[54]. These approvals also provide promising research directions for more diseases [Table 1].
FDA-approved mRNA-LNP on the market
| Drug Name | Brand Name | Disease | Manufacturer | Approval Year | LNP Composition |
| Patisiran | Onpattro | hATTR | Alnylam Pharmaceuticals | 2018 | DLin-MC3-DMA (cationic lipid), DSPC, cholesterol, PEG2000-DMG |
| BNT162b2 | Comirnaty | COVID-19 | Pfizer/BioNTech | 2020 | ALC-0315 (cationic lipid), DSPC, cholesterol, ALC-0159 (PEG-lipid) |
| mRNA-1273 | Spikevax | COVID-19 | Moderna | 2020 | SM-102 (cationic lipid), DSPC, cholesterol, PEG2000-DMG |
| mRNA-1345 | mRESVIA | RSV | Moderna | 2024 | SM-102 (cationic lipid), DSPC, cholesterol, PEG2000-DMG |
mRNA-LNP FOR PULMONARY DISEASES
Building on the advantages of LNPs as an efficient mRNA delivery system, their potential for treating pulmonary diseases is increasingly recognized. Pulmonary diseases encompass a range of conditions, including viral infections, non-viral infections, and lung cancer, each with distinct pathophysiological mechanisms and therapeutic challenges. As a flexible therapeutic platform, mRNA-LNPs offer significant promise in addressing these diverse challenges, providing tailored solutions for targeted treatment strategies [Figure 2].

Figure 2. mRNA-LNP for pulmonary diseases. LNP: Lipid nanoparticle; IPF: idiopathic pulmonary fibrosis; RSV: respiratory syncytial virus.
Viral infection
Viral infections of the lungs are a major cause of respiratory diseases[55]. They progress through viral invasion of host cells, where the transcription and translation machinery is hijacked for replication, resulting in tissue damage and systemic complications[56]. However, traditional antiviral therapies face significant challenges, particularly their limited efficacy against rapidly mutating viruses. Due to its efficient delivery and flexible design capabilities, mRNA-LNP technology has emerged as a promising therapeutic strategy for viral respiratory infections. This section describes the application of the mRNA-LNP system in viral respiratory diseases in terms of influenza virus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and RSV[47].
Influenza virus
Influenza virus, a highly mutable RNA virus, infects host cells by binding to sialic acid receptors through hemagglutinin (HA) proteins on its surface. Following entry into host cells, the virus replicates in the nucleus, causing acute respiratory infections and inflammation[57]. Frequent antigenic drift undermines the effectiveness of conventional vaccines and antiviral drugs. However, mRNA-LNP-based therapies offer a groundbreaking solution[58]. By encoding conserved regions of viral proteins, mRNA vaccines stimulate the production of specific antibodies[47]. For instance, the mRNA-1440 and mRNA-1851 vaccines, targeting the H10N8 and H7N9 influenza strains, demonstrated high immunogenicity in Phase I clinical trials (NCT03076385, NCT03314504)[59,60]. The clinical data of the H10N8 mRNA vaccine demonstrated that 100% of participants achieved hemagglutination inhibition (HAI) titers ≥ 40 and 87% reached micro-neutralization (MN) titers ≥ 20, along with an acceptable safety profile and no serious adverse events[59]. Additionally, Magini et al.[61] developed an RNA vaccine based on self-amplifying mRNA (SAM) encapsulated in LNPs, which enhanced CD4+ and CD8+ T cell recruitment. This vaccine induced robust T cell responses, with NP-specific CD8+ T cell frequencies reaching 1%-2%, significantly reducing lung viral titers by more than 10-fold. Moreover, there are multiple novel mRNA-LNPs targeting different mid-influenza viruses such as H1N1, H5N1, and H10N8 + H7N9, collectively demonstrating their potential as emerging therapeutics[59,62,63].
SARS-CoV-2
SARS-CoV-2 invades host cells by binding its spike (S) protein to the angiotensin-converting enzyme 2 (ACE2) receptor[64], causing severe lung damage such as acute lung injury (ALI) and acute respiratory distress syndrome (ARDS)[65]. The application of mRNA-LNP technology in COVID-19 vaccine development has been revolutionary. Pfizer-BioNTech and Moderna mRNA vaccines utilize LNPs to deliver mRNA encoding the SARS-CoV-2 spike protein, successfully inducing specific neutralizing antibodies and significantly reducing infection rates and severity[66,67]. Additionally, the rapid adaptability of mRNA technology enables swift adjustments to address emerging viral variants. Chen et al.[68] developed a novel ionizable lipid-based LNP (4N4T) platform to deliver mRNA encoding SARS-CoV-2 receptor-binding domain (RBD) antigens. This platform elicited robust adaptive immune responses against Delta and Omicron variants, with higher RBD-specific IgG and neutralizing antibody titers. Focusing on various directions such as non-structural proteins and Omicron variants, mRNA-LNP has made significant contributions to human health[69-78].
mRNA vaccines have played a transformative role in the fight against SARS-CoV-2, demonstrating high efficacy and rapid scalability in clinical applications. The two widely used mRNA vaccines, BNT162b2 (Pfizer-BioNTech) and mRNA-1273 (Moderna), showed 95% and 94.1% efficacy, respectively, against symptomatic COVID-19 infection in Phase III clinical trials[79,80]. Dangan et al.[81] also support the vaccine effectiveness, with an estimated effectiveness of 72% for preventing COVID-19 deaths within 14 to 20 days after the first injection. The adaptability of mRNA technology has facilitated the rapid update of vaccines to address emerging variants, which is important in future pandemic response and preparedness[82].
RSV
RSV is a leading cause of lower respiratory tract infections in infants and young children, primarily infecting cells through the interaction between its F protein and host cell receptors[83,84]. mRNA-LNP-based RSV vaccines have demonstrated significant advantages[85]. For example, Wu et al.[86] designed a bivalent vaccine encoding RSV F protein and SARS-CoV-2 Omicron spike protein. This vaccine successfully induced specific antibody and cellular immune responses in BALB/c mice models [Figure 3]. In addition, mRNA-1345, an mRNA-based RSV vaccine developed by Moderna, has demonstrated promising tolerability and immunogenicity in clinical trials[54].

Figure 3. Design of an mRNA vaccine candidate against RSV and SARS-CoV-2[86]. RSV: Respiratory syncytial virus; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2. SF-LNP: Lipid nanoparticle which delivered mRNAs encoding SARS-CoV-2 Omicron spike (S) and RSV fusion (F) proteins simultaneously;S-LNP: lipid nanoparticle which delivered mRNAs encoding SARS-CoV-2 Omicron spike (S) proteins; F-LNP: lipid nanoparticle which delivered mRNAs encoding RSV fusion (F) proteins.
Non-infectious lung disease
In addition to viral infections, lung diseases also include non-infectious diseases such as idiopathic pulmonary fibrosis[87] and vascular endothelial dysfunction[88]. The pathogenesis of these diseases is complex, and it is difficult to be cured effectively by existing therapies. In recent years, mRNA-LNP technology has offered them a new therapeutic option due to its efficient delivery and precise gene regulation capability[89].
Idiopathic Pulmonary Fibrosis (IPF)
IPF is a lethal interstitial lung disease characterized by abnormal repair of alveolar epithelial cells after injury[90,91]. This process is accompanied by fibroblast activation and excessive extracellular matrix (ECM) deposition, leading to a progressive decline in lung function. mRNA-LNP therapy delivers functional mRNA targeting fibroblasts and alveolar epithelial cells[92-94]. This provides new insights into IPF precision therapy. For example, Massaro et al.[95] established a small mRNA-LNP delivery system with a high encapsulation efficiency and stable mRNA release properties. In lung fibrosis models, the mRNA targeted the alveolar region and induced protein expression, successfully inhibiting the fibrotic process. Additionally, Wang et al.[96] developed an mRNA-LNP-based inhalation therapeutic strategy, which restored the stemness of Alveolar Type 2 cells and promoted alveolar regeneration by delivering mRNAs encoding cytochrome b5 reductase 3 (CYB5R3) and bone morphogenetic protein 4 (BMP4) [Figure 4]. Although mRNA-LNP therapy for IPF has just been proposed, the feasibility of mRNA-delivered therapy was also demonstrated previously by mRNA nano-formulations to enhance matrix metallopeptidase-13 (MMP-13) expression[97].

Figure 4. mRNA -LNP reversal model of pulmonary fibrosis in mice[96]. CYB5R3: Cytochrome b5 reductase 3; BMP4: bone morphogenetic protein 4; ECM: excessive extracellular matrix; LNP: lipid nanoparticle. TGF: Transforming growth factor; NADH: nicotinamide adenine dinucleotide (reduced form); NAD+: nicotinamide adenine dinucleotide (oxidized form); SAAP: senescene-associated secretory phenotype; AT1: alveolar type 1 epithelial cells.
Endothelial cell dysfunction
Characterized by impaired endothelial cell (EC) barrier function, vascular endothelial dysfunction (VED) is an important pathomechanism in several lung diseases, leading to vascular leakage and inflammatory cell infiltration[98-100]. mRNA-LNP technology offers innovative therapeutic options for VED by delivering mRNAs encoding endothelial protective proteins. A study developed an mRNA-LNP system delivering mRNA encoding the Tie2 agonist COMP-Ang1. In a mouse model of acute lung injury, Tie2 signaling pathway activation restored endothelial barrier integrity and reduced the inflammatory response, effectively decreasing pulmonary vascular leakage and neutrophil infiltration[101]. Meanwhile, Zhao et al.[102] developed a lipid nanoparticle targeting pulmonary endothelium for lung-LNP (LuLNP). LuLNP ameliorated the impaired endothelial cell phenotype associated with influenza injury by encapsulating Vegfa mRNA, a downstream signaling factor for TGF-βR2. These newly developed mRNA-LNP technologies demonstrate the potential for treating vascular endothelial dysfunction.
Lung tumor
Lung tumors include a wide range of benign and malignant diseases, such as lymphangioleiomyomatosis and lung cancer. They have different pathogenic mechanisms and characteristics, but all of them are challenged by drug resistance and poor prognosis[103]. Recently, mRNA-LNP has emerged as a promising approach for precise and personalized treatment of lung tumors.
Lymphangioleiomyomatosis (LAM)
LAM is a rare lung disease characterized by the abnormal proliferation of smooth muscle-like cells (LAM cells) in the lungs and lymphatic vessels[104]. It is a genetic disease with mutations in the tuberous sclerosis complex (TSC1/TSC2) gene that activates the mammalian target of the rapamycin (mTOR) pathway[105]. Based on the severe side effects of the current mTOR1 inhibitor rapamycin (simethicone)[106], Qiu et al.[107] evaluated the therapeutic availability of lung-targeted LNP delivery of Tsc2 gene mRNA. Results showed that the LNP model effectively targeted Tsc2 mRNA delivery to Tsc2-null, kidney-derived epithelial tumor cells and significantly inhibited tumor cell growth [Figure 5]. This provides critical evidence for mRNA-LNP in the LAM treatment.

Figure 5. mRNA-LNP delivery for LAM treatment[107]. (A) Schematic of hybrid lipid hLNPs; (B) In vivo fluorescence imaging demonstrating EGFP expression localized in mouse LAM lungs following intravenous injection of EGFP mRNA-loaded hLNPs; (C) Confocal immunofluorescence images showing colocalization of phospho-S6 and EGFP, confirming targeted delivery to tumor cells. LNP: Lipid nanoparticle; LAM: lymphangioleiomyomatosis; DMG-PEG: 1,2-dimyristoyl-rac-glycero-3-methoxy-polyethylene glycol; EGFP: enhanced green fluorescent protein; hLNP: hybrid lipid nanoparticle; pS6: phosphorylated ribosomal protein S6.
Lung cancer
Lung cancer, a highly heterogeneous and aggressive malignant lung tumor, is the disease with the highest cancer-related mortality[7]. Its pathogenesis usually involves aberrant cell proliferation caused by mutations in driver genes, accompanied by remodeling of the tumor microenvironment and immune escape, leading to rapid tumor growth and metastasis[108]. Existing treatments include surgery, chemotherapy, radiotherapy, targeted therapies, and immune checkpoint inhibitors, but their efficacy is limited by tumor resistance and individual patient differences[109]. Therefore, there is an urgent need to find new therapeutic modalities, and mRNA-LNP has become a research hotspot[110]. For example, Cheng et al.[111] designed G3139-GAP-LNPs with a high encapsulation rate based on Bcl-2, an anti-apoptotic gene that is overexpressed in cancers. It effectively downregulated the expression of bcl-2 in A549 cells, inhibited tumor growth, and prolonged the survival period. This technology was also further proved to be effective in the T7 peptide-conjugated lipid nanoparticles[112]. In addition, Yu et al.[113] developed LNPLung, a lipid nanoparticle system for targeted delivery of IL-15 superagonist mRNA to lung tissue. In a lung tumor model, LNPLung effectively stimulated CD8+ T and NK cells, inhibited tumor growth, and reduced cytokine leakage. This successfully enhanced immune activation in response to tumors while reducing systemic toxicity to prevent damage to normal tissues. In addition, non-human primate models further validated its safety with high specificity and low off-target rates, which demonstrates the strong clinical potential of LNP for lung cancer. Moreover, mRNA-targeted lung cancer therapy is a promising way to cure patients in the future, even if LNP delivery is not used. The BNT116 mRNA vaccine against the lung cancer subtype non-small cell lung cancer (NSCLC) has been shown to be clinically effective (NCT05142189), with a good safety profile and promising anti-tumor activity[114]. Therefore, the development of mRNA-LNP is a new dawn for the treatment of lung cancer.
Although mRNA-LNP has great potential in both lung cancer and lung infections, the requirements for them in these diseases are quite different. Infectious diseases such as influenza virus, SARS-CoV-2, and RSV require fast-acting vaccines to stimulate immune responses or to replace defective proteins. At the same time, they are highly demanding in terms of effectively entering host cells and overcoming cellular barriers. A formulation that ensures rapid cellular uptake and effective antigen presentation is therefore required[47]. In contrast, the continuous growth of cancer cells and their superior immune escape place a higher demand on the dosing, persistence, and high targeting of mRNA-LNPs to avoid inadvertent injury to normal cells. This makes nanocarriers require more rigorous design, such as specific targeting ligands for effective drug delivery[31,115]. The different characteristics of the disease determine the need for the drug. These distinctions highlight the necessity for tailored mRNA-LNP designs, optimizing lipid composition, particle size, and targeting ligands to meet the unique pharmacokinetic and immunological demands of each pulmonary condition[46,116]. As mRNA therapeutics advance, the refinement of LNP formulations will be essential in maximizing their potential across diverse lung diseases.
TARGETING STRATEGY OF mRNA-LNP PULMONARY DELIVERY
As mentioned above, the lungs are important in maintaining body homeostasis. Many short-term and long-term health problems can damage the lungs. These include diseases like cystic fibrosis, severe breathing problems (ARDS), inherited lung disease, scarring of the lungs, cancer, and infections caused by bacteria and viruses. Accordingly, due to the lack of effective and widely available drug treatments and limitations in clinical care, mRNA-based gene therapy shows strong development potential for these diseases. In this review, we summarize reported mRNA-LNP strategies for lung-targeted delivery as a reference for researchers in the field.
The most common delivery route for LNPs is systemic administration, which often results in hepatic accumulation. To this day, many researchers are focused on enabling LNPs with extra-hepatic targeting capabilities. Meanwhile, considering the unique physiological structure of the lungs, an effective targeting strategy is to employ respiratory tract approaches for drug delivery, including nebulized inhalation, intratracheal administration, and intranasal administration. Here, we review the developed lung-targeting LNP strategies, categorized by different administration routes [Figure 6].
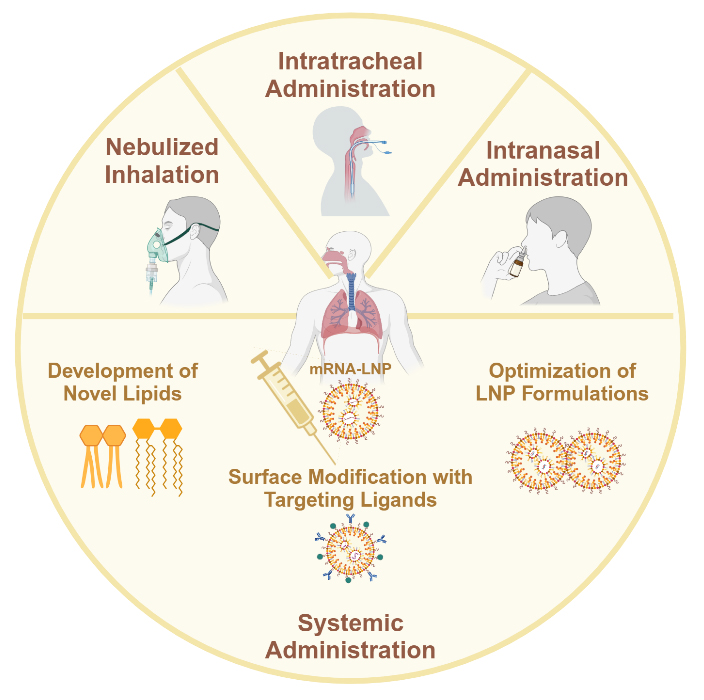
Figure 6. Different administration routes for mRNA-LNP pulmonary delivery. LNP: Lipid nanoparticle.
Nebulized inhalation
During nebulization, drug solutions are converted into aerosol droplets for respiratory delivery[117]. Through nebulized inhalation, aerosol droplets travel directly to the airway and deposit throughout the respiratory tract[118]. Among the various routes for administering mRNA-LNPs, nebulized inhalation offers significant advantages: minimal systemic exposure, low side effects, avoidance of hepatic and renal clearance, and no invasive injections[119].
However, delivery of mRNA-LNPs through nebulized inhalation faces a significant challenge: the nebulization process exerts shear stress on the LNPs, leading to their instability, aggregation, and rupture[120]. To enhance LNP stability during nebulization, researchers are focusing on optimizing LNP formulation strategies, which is crucial for maintaining the structural integrity of particles [Figure 7].

Figure 7. Nebulized Inhalation. Reprinted with permission from[121]. Copyright 2025 American Chemical Society. LNP: Lipid nanoparticle.
Since LNPs were initially developed for systemic delivery, most optimization work is based on systemic administration. However, LNP design rules for nebulized delivery could be very different, which created a need to establish new design principles for LNPs specifically intended for nebulization systems. Lokugamage et al.[62] developed a cluster-based iterative screening method for optimizing LNP formulation. Their research showed that for nebulized inhalation delivery, the concentration of PEG lipids in LNPs is crucial for effective pulmonary delivery, and LNPs with high molar ratios of PEG and cationic lipids demonstrated superior lung delivery performance.
After that, Kim et al.[121] enhanced the shear resistance and mucus penetration of LNPs by increasing PEG lipid. Meanwhile, they added β-sitosterol (a cholesterol analog) to provide LNPs with a polyhedral shape that facilitates endosomal escape. The optimized LNPs enhanced mRNA transfection efficiency and achieved localized protein production in mouse lungs after inhalation, showing no signs of pulmonary or systemic toxicity.
It should be noted that nebulization is a complex process, so many key factors besides the LNP formulation design could also affect the stability of LNPs during nebulization, such as buffer conditions and excipient selection.
Jiang et al.[122] evaluated and screened mRNA-LNP pulmonary delivery based on fully differentiated primary lung epithelial cells cultured at the air-liquid interface, avoiding the inherent nonlinear effects of nanoparticle pools. They identified a final combination of ionizable lipids, charge-stabilizing formulation, and stability-enhancing excipients that improved the stability of LNPs after nebulization, significantly enhancing pulmonary mRNA delivery.
Similarly, Bai et al.[123] designed a “LOOP” platform with a four-step workflow for developing inhaled LNPs specifically for pulmonary mRNA delivery, which enabled researchers to obtain LNPs with superior shear force resistance and high mRNA expression without requiring extensive screening [Figure 8].
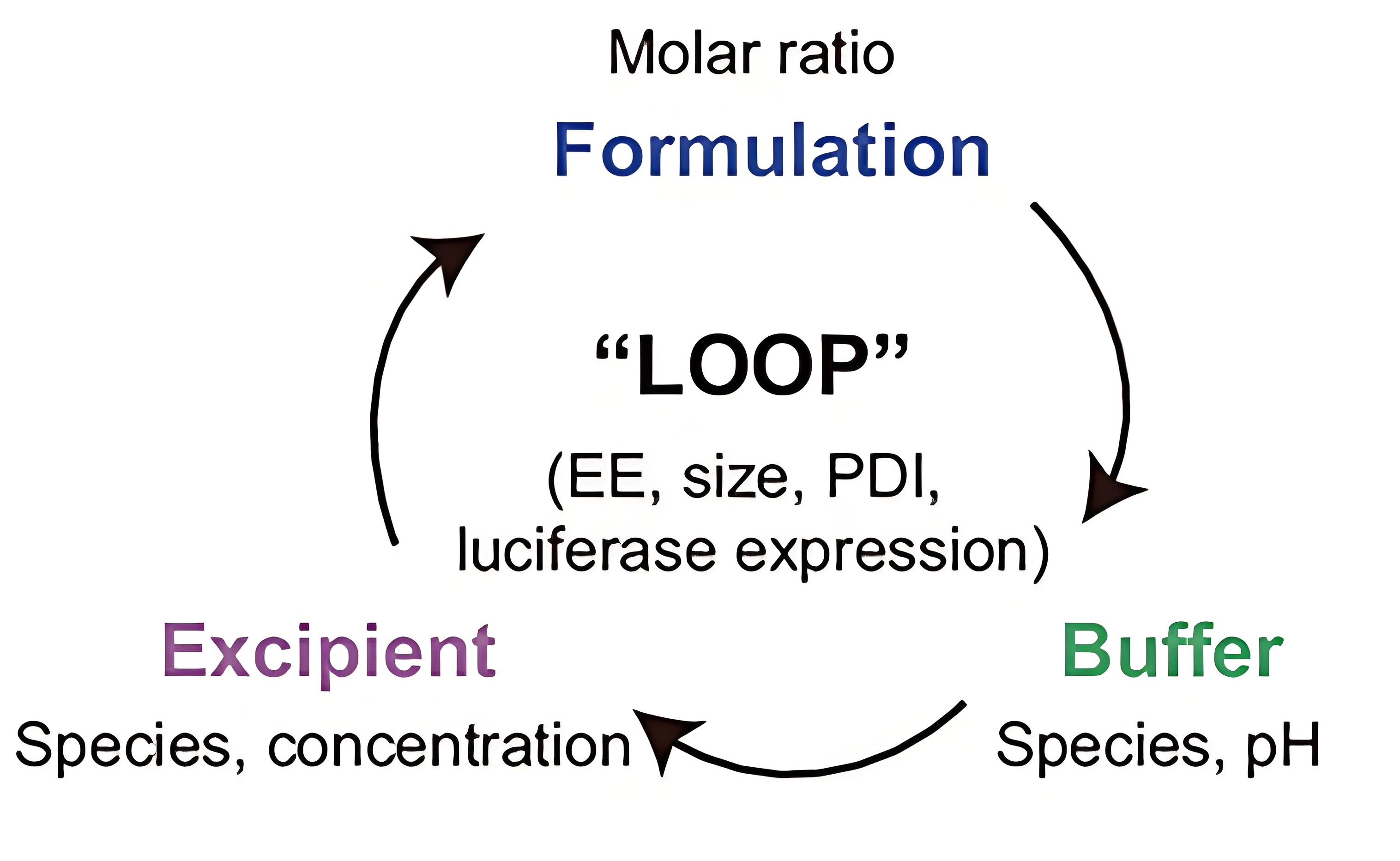
Figure 8. Screening of inhaled LNP for nebulized mRNA delivery[123]. LNP: Lipid nanoparticle; EE: encapsulation efficiency PDI: polymer dispersity index.
Subsequently, another study pointed out that although positively charged LNPs could remain stable during nebulization and effectively deliver mRNA to the lungs, they primarily transfect lung epithelial cells, which are not ideal for delivering mRNA vaccines. Instead, they developed a charge-assisted stabilization (CAS) strategy by adding negatively charged peptide-lipid conjugates into the conventional four-component LNP[124]. This approach could induce electrostatic repulsions between LNPs and enhance their colloidal stability. Most importantly, mRNA-CAS-LNP demonstrated effective transfection on dendritic cells within the lungs following inhalation, establishing it as a promising candidate for mRNA vaccine delivery.
Furthermore, recent research has shown that coating nanoparticles with lung surface components enhances their diffusion across the mucus barrier through interfacial delivery, enabling them to penetrate deep lung tissue and enter epithelial cells more effectively. Building on this insight, Wang et al.[96] created pulmonary surfactant-biomimetic LNPs by adding l-alpha-Dipalmitoyl phosphatidylcholine (DPPC) (a phospholipid abundant in lung surfactant) into conventional lipid components. They also developed a class of ionizable lipids based on γ-aminobutyric acid (GABA) and optimized the formulation of LNPs for high-efficiency mRNA delivery through in vitro screening. Optimized mRNA-LNPs could markedly reduce lung fibrosis after inhalation.
Intratracheal administration of mRNA-LNPs
Expression kinetics studies have shown that the administration route of mRNA-LNPs could affect where and when proteins are expressed in the body. Considering the specific structure of the respiratory system, intratracheal administration could effectively deliver nanoparticles to the lung[125,126]. Meanwhile, the small size of LNPs allows them to adhere to the mucosal surface, prolonging their retention time in the airways[127,128]. Currently, many studies have investigated the intratracheal administration of mRNA-LNPs.
Intratracheal administration based on nebulization
Stable nebulization of mRNA-LNPs could facilitate their intratracheal administration. Zhang et al.[129] developed a design of experiments (DOE) strategy to screen LNP formulations for mRNA delivery to the lungs, which could streamline the formulation screening process [Figure 9]. Through this approach, they identified four formulations with enhanced intracellular protein expression after aerosolization and showed strong luciferase expression in mouse lungs. Their research showed that LNPs could effectively deliver mRNA to the lungs when administered intratracheally following nebulization.

Figure 9. Schematic of the DOE strategy to find LNP formulations for mRNA delivery to the lungs following intratracheal administration after nebulization[129]. DOE: Design of experiments; LNP: lipid nanoparticle.
Intratracheal administration based on dry powder
Despite their widespread use, nebulizers and meter dose inhalers present challenges with patient compliance. To address these limitations, dry powder inhalers (DPIs) were developed. These devices offer several advantages: they are more affordable, portable, propellant-free, breath-activated, and simple to use. Additionally, the solid dispersed state of dry powder products (DPP) can improve the stability of mRNA-LNPs. Kriis et al.[130] successfully achieved the first intratracheal delivery of mRNA-LNP using solid spray-dried formulations. In this proof-of-concept study, they substituted lipids of LNPs to temperature-resistant variants, which could maintain functionality after spray drying and reconstitution in the water and realize effective mRNA delivery both in vitro and in vivo [Figure 10].

Figure 10. Spray-dried LNPs enable intratracheal delivery of mRNA[130]. LNP: Lipid nanoparticle.
After that, Sarode et al.[131] also developed dry powder formulations of mRNA-LNPs suitable for inhalation. They optimized these formulations by evaluating different solvents, lipid/mRNA concentrations, processing parameters, and LNP compositions. Their work demonstrates that optimized mRNA-LNPs administered as DPPs could achieve effective functional pulmonary delivery, showing the potential for developing mRNA-LNP dry powder formulations for lung delivery.
Intratracheal administration based on solution
On the other hand, although nebulization technology has been widely used for pulmonary drug delivery, some studies suggest that nebulization may disrupt the structure of LNPs, thereby affecting their actual efficacy[132]. Intratracheal administration of mRNA-LNP solutions into mice and allowing delivery to the lungs through inhalation create an opportunity to address this limitation.
Research by Pardi et al.[133] has shown that intratracheal administration of mRNA-LNPs could achieve high protein expression in the lungs. In another study, Massaro et al.[95] used a bleomycin mouse model of lung fibrosis, where intratracheal administration of mRNA-LNP enabled site-specific lung accumulation [Figure 11]. Notably, 24 h after administration, no mRNA-related signals were detected in organs other than the lungs, highlighting the superiority of intratracheal injection for lung-targeted delivery.

Figure 11. Schematic of LNPs for delivery of mRNA to lungs undergoing fibrosis following intratracheal administration[95]. LNP: Lipid nanoparticle. DPPC: l-alpha-Dipalmitoyl phosphatidylcholine; DOTAP: 1,2-dioleoyl-3-trimethylammonium-propane; DMA: dimethylamine; DSPE: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine; PEG: polyethylene glycol; AEC: aminoethyl carbazole.
In further studies, Geng et al.[134] tried to increase the protein expression in the lungs with intratracheal administration by adjusting the lipid composition of LNPs. Their investigation revealed that protein expression could be significantly enhanced by decreasing the PEG molarity, using DSG-PEG as PEGylated lipids, and replacing DSPC with DOPE [Figure 12]. These findings contribute valuable insights to the ongoing development of mRNA-LNPs for intratracheal administration.

Figure 12. mRNA-LNP had robust protein expression in the lungs of mice following intratracheal administration[134]. LNP: Lipid nanoparticle.
Intranasal administration of mRNA-LNPs
Similarly, while nebulization can deliver LNPs specifically to the lungs, this process subjects particles to shear forces that might increase their size and decrease their encapsulation efficiency. Therefore, Tam et al.[135] developed an LNP formulation for intranasal delivery to avoid damage from nebulization [Figure 13]. They administered mRNA-LNP intranasally by dripping it into a single mouse nostril, allowing natural inhalation. The authors developed a small library of six LNP formulations using different helper lipid compositions. In in vivo studies, when LNPs containing helper lipids [1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), DOPC, egg sphingomyelin (ESM) or dioleoylphosphatidylserine (DOPS)] with luciferase mRNA were delivered intranasally, they could produce higher luminescence levels in the nasal cavity and lungs. These results demonstrated that LNP-packaged mRNA could be administered intranasally to target the airway epithelium for potential therapeutic applications.

Figure 13. Lipid nanoparticle formulations for optimal RNA-based topical delivery to murine airways[135]. GFP: Green fluorescence protein; DiD: 1,1'-dioctadecyl-3,3,3',3'-tetramethylindodicarbocyanine,4-chlorobenzenesulfonate salt; DSPC:1,2-distearoyl-sn-glycero-3-phosphocholine; DOPC: dioleoylphosphatidylcholine; DOPE: dioleoylphosphatidylethanolamine; ESM: egg sphingomyelin; DOPG: egg sphingomyelin; PEG: polyethylene glycol; LNP: lipid nanoparticle.
Systemic administration of mRNA-LNPs
After systemic administration, conventional four-component LNPs predominantly accumulate in liver and spleen tissues[136]. To this day, researchers have undertaken extensive investigations to develop organ-specific LNP delivery systems. Current approaches could be classified into three strategies: developing novel lipids, optimizing LNP formulations, and modifying with targeting ligands.
Development of novel lipids
With the increased understanding of LNP in vivo delivery processes, there is a consensus that when LNPs enter the body, their outer surface is rapidly covered with plasma proteins, which are called protein corona[137,138]. The protein corona reshapes the surface properties of LNPs and significantly affects their interactions with organs and cells[139-141]. Therefore, by modifying the lipid structure within LNPs, researchers can alter the types of plasma proteins in the protein corona, thereby achieving targeted delivery of LNPs to specific organs.
Qiu et al.[107] synthesized a library of amide bond-containing lipidoids for N-series LNPs, and their work demonstrated that N-series LNPs could distribute exclusively to lung tissue after systemic administration. Furthermore, by modifying the linker structure in the lipid tails, N-series LNPs could achieve targeting of different lung cell subpopulations. Subsequent proteomic analysis revealed that this organ selectivity is due to the phospholipid tail structure potentially having an affinity for specific serum proteins, which determines the composition of the protein corona and ultimately controls the fate of LNPs [Figure 14].

Figure 14. Synthesis and in vivo screening of N-series LNPs[107]. LNP: Lipid nanoparticle.
In another study, a library of 570 ionizable lipidoids with different branched tails was synthesized, and a novel branched-tail LNP was screened out, which possessed the unique ability to selectively transfect immune cells in the lung without using cationic helper lipids[142].
In addition, some thiolipid-based ionizable lipids have also been reported to have lung-targeting potential. Eygeris et al.[143] reported the identification and evaluation of ionizable lipids containing thiophene moieties (Thio-lipids). They synthesized 47 thio-lipids and optimized the formulation using high-throughput formulation and barcoding technology, and lipid 29d LNP could achieve dual targeting to the lung and spleen [Figure 15]. Similarly, Popoola et al.[89] reported the synthesis of two sulfonium lipids, DHSEH and DOSEH. Sulfonium lipid nanoparticles (sLNPs) with these lipids could effectively and safely deliver various types of mRNA to the lungs through intravenous administration and achieve high protein expression.

Figure 15. Synthesis and in vivo screening of N-series LNPs[143]. DMG-PEG: 1,2-Dimyristoyl-rac-glycero-3-methoxy-polyethylene glycol; DOPC: dioleoylphosphatidylcholine; LNP: lipid nanoparticle; PBS: phosphate buffered saline.
Moreover, several other novel ionizable lipids have been developed to enhance the pulmonary delivery of mRNA-LNPs, including the piperazine‐based biodegradable ionizable lipid (244cis)[144], ionizable amino-polyesters (APEs)[145], and imidazolium IM21.7c[146].
Although most researchers have focused on developing ionizable cationic lipids, modifying other LNP components could also achieve lung-targeted delivery. Here, Zeng et al.[147] developed a cationic lipid pair (CLP) strategy that redirects LNPs from the liver to the lungs [Figure 16]. In this strategy, phospholipids in the traditional liver-targeting LNPs were replaced by their ionizable lipid-derived quaternary ammonium lipid counterpart, which could significantly enhance the pulmonary delivery of mRNA-LNPs in vivo.

Figure 16. CLP strategy to adjusting liver-to-lung tropism of LNPs for in vivo Mrna delivery[147]. CLP: Cationic lipid pair; LNP: lipid nanoparticle; DMG-PEG: 1,2-dimyristoyl-rac-glycero-3-methoxy-polyethylene glycol.
Optimization of LNP formulations
Daniel J Siegwart’s team pioneered a selective organ targeting (SORT) strategy, which could rationally design LNPs for extra-hepatic mRNA delivery via intravenous administration. In this strategy, a fifth component (SORT molecule) is added to the traditional four-component LNP, which could change the liver-targeting properties of LNPs in vivo. Initial research demonstrated that incorporating the cationic lipid 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) as a SORT molecule facilitated targeted delivery to lung tissue[148]. As the amount of DOTAP increased, the site of protein expression shifted markedly from the liver to the spleen and ultimately to the lungs, demonstrating a clear organ-specific distribution pattern. Subsequently, researchers tested a series of permanent cationic lipids as SORT molecules and optimized their incorporation ratios of LNPs[149]. This led to the development of second-generation Lung SORT LNPs (DOTAP40 LNPs), which showed exceptional effectiveness in pulmonary mRNA delivery. Building upon this foundation, researchers conducted a comprehensive screening of structurally diverse cationic lipids as SORT molecules, and conducted in-depth research on how the chemical structure of SORT molecules affects the formation of protein corona in vivo[150]. Finally, their recent research demonstrated that SORT LNPs achieve high levels of genome editing in lung stem cells, with effects persisting for over 1.8 years[151]. In a word, the SORT strategy enables LNPs to overcome the delivery barrier of hepatocyte accumulation and target the lungs. Its endogenous targeting mechanism relies on the protein corona formed on LNPs during in vivo delivery. Specifically, SORT molecules exposed on the LNP surface after the desorption of PEG lipids could bind to different plasma proteins, and these surface-bound proteins will interact with homologous receptors highly expressed in specific tissues, thereby mediating the organ-targeting properties of LNPs [Figure 17].

Figure 17. SORT nanoparticles for tissue-specific mRNA delivery[152]. SORT: Selective organ targeting.
Inspired by the SORT strategy, other studies have also reported lung-targeted delivery of LNPs through the addition of extra cationic lipids. Wan et al.[71] obtained cationic targeting lipids by converting ionizable lipids to their quaternary ammonium salt form through a reaction with methyl iodide. After adding this targeting lipid and replacing the helper lipid DSPC with DOPE, they obtained lung-selective LNPs, which were capable of precisely delivering mRNA to the lungs [Figure 18].

Figure 18. Lung-selective lipid nanoparticle systems[71]. (A) Structural formula and schematic diagram of each component of lung-selective and liver-selective LNPs; (B) Schematic diagram of lung-selective and liver-selective LNPs. (C) TEM results: upper image (Lung-LNPs) and lower image (Liver-LNPs). LNP: Lipid nanoparticle. DOPE: dioleoylphosphatidylethanolamine; DSPC: 1,2-distearoyl-sn-glycero-3-phosphocholine; PEG: polyethylene glycol; TEM: transmission electron microscopy.
Although the SORT strategy demonstrates efficacy, it introduces additional complexity to the formulation, which may extend regulatory review timelines and present challenges in the manufacturing process. Therefore, some studies have focused on simplifying LNP composition while maintaining shifts in tropism.
LoPresti et al.[153] demonstrated that replacing the standard helper lipid DOPE with an alternative cationic lipid like DOTAP could cause a pronounced shift of LNP specificity to the lungs, while maintaining a four-component formulation.
Another study found this charge-dependent tropism depends on the composition of cationic lipids. Using cationic cholesterol alone would still localize LNPs to the liver, while using both cationic cholesterol and cationic helper lipids could enhance LNP targeting to the lungs[154].
On the other hand, reducing LNP components might increase mRNA delivery efficiency. Fei et al.[155] developed a three-component LNP and integrated unique microRNA target sites into the mRNA scaffold, achieving precise delivery of mRNA to tumor cells in the lungs. They named this approach SELECT (Simplified LNP with Engineered mRNA for Cell-type Targeting). This innovative method opens new possibilities for mRNA therapy that can target both specific organs and cell types simultaneously.
Surface modification with targeting ligands
The surface modification of targeting ligands could allow LNPs to reach and interact with specific cell types or tissues. Plasmalemma vesicle associated protein (PV1) is a known caveolae-associated protein that mediates internalization independent of clathrin/dynamin pathways[156]. Given that caveolae make up over 70% of the lung capillary endothelial membrane[157], PV1 presents a promising target for delivering LNPs to the lungs. Here, Li et al.[158] covalently conjugated the PV1 antibody to the surface of LNPs, which could effectively target mRNA-LNPs to the lung tissue and increase protein expression levels in the lungs by 40-fold [Figure 19].

Figure 19. Anti-Plasmalemma vesicle associated protein-modified mRNA-LNPs targeted the lungs[158]. LNP: Lipid nanoparticle.
CONCLUSIONS AND FUTURE PERSPECTIVES
Nowadays, administration routes of lung-targeted mRNA-LNP fall into two categories: respiratory administration (including inhalation, intratracheal administration, and intranasal administration) and systemic administration (intravenous injection). The main advantage of respiratory administration lies in its ability to deliver mRNA-LNP directly to the lungs, avoiding losses during systemic circulation. This significantly improves mRNA delivery efficiency to the lungs, thereby requiring lower doses and reducing systemic side effects. However, respiratory administration also has drawbacks, including complex delivery procedures, poor patient compliance, and potential failure to reach deep lung tissues. Current research focuses on simplifying devices for inhalation and enhancing LNP stability through nebulization.
In contrast, while systemic administration faces challenges with first-pass effects and off-target distribution, it also offers several benefits. The formulation of mRNA-LNP needs no special handling, and its administration is simple, convenient, and has high patient compliance. Moreover, optimized LNPs could achieve specific targeting of diverse cell populations and deeper lung distribution. In summary, each administration route presents unique advantages and limitations for lung-targeted mRNA delivery. The optimal choice depends on specific therapeutic objectives, disease characteristics, and individual patient factors.
Given the current state of research and applications, future research trends in mRNA LNPs targeting the lungs may involve the following areas. First, to develop an intelligent response LNP system based on the inherent physiological characteristics of the lung to improve the targeting efficiency of LNP in the lung. For example, the design based on respiratory mechanical response can mimic the mechanical force of alveolar expansion/contraction and achieve respiration-triggered slow drug release by mimicking the structure of alveolar epithelial folds or the tidal respiratory rhythm to control the pore opening/closing system, thereby enhancing the retention efficiency of the drug in deep lung tissues. Such designs are expected to overcome the limitations of traditional carriers in adapting to complex physiological environments. Second, bionic design is used to improve the precision of targeting strategies. For example, modification by exosomal membrane wrapping or synthetic lung surface active protein D optimizes the air-liquid interface penetration ability of LNP and reduces macrophage phagocytosis. Third, to improve the stability of the LNP structure. Since pulmonary delivery involves multiple barrier challenges such as nuclease degradation, high-temperature nebulization and mucus removal, LNP delivery systems can be designed with multiple layers of protection - for example, an inner core with ionic chelators to stabilize the mRNA structure, an intermediate layer with heat stable lipids to withstand nebulization, and an outer layer coated with amphoteric ionic polymers to reduce mucus adsorption and prolong retention time. Additionally, future directions for LNPs include reducing the toxicity of LNPs in addition to further improving delivery efficiency and stability. Cationic lipids with permanent positive charge can be toxic to cells, and future LNPs may require screening for low-toxicity lipid components and optimization of surface charge distribution, which may significantly reduce cytotoxicity and immunogenicity.
In conclusion, targeted mRNA LNPs for lung disease therapy are still in their initial stages, and more research efforts are needed in the future. With the development of technology, LNPs are moving toward intelligence, precision, and safety, and their breakthroughs will drive the precision treatment of respiratory diseases to a new stage.
DECLARATIONS
Authors’ contributions
Conceived and designed the article: Luo, L.; You, J.; Yang, F.
Drafted and revised manuscript: Liu, X.; Ji, S.
Assistance with manuscript drafting: Cai, Y.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Financial support and sponsorship
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Murray, J. F. The structure and function of the lung. Int. J. Tuberc. Lung. Dis. 2010, 14, 391-6.
2. Weibel, E. R. Lung morphometry: the link between structure and function. Cell. Tissue. Res. 2017, 367, 413-26.
3. Pettigrew, M. M.; Tanner, W.; Harris, A. D. The lung microbiome and pneumonia. J. Infect. Dis. 2021, 223, S241-5.
4. Mokrá, D. Acute lung injury - from pathophysiology to treatment. Physiol. Res. 2020, 69, S353-66.
5. Alfahad, A. J.; Alzaydi, M. M.; Aldossary, A. M.; et al. Current views in chronic obstructive pulmonary disease pathogenesis and management. Saudi. Pharm. J. 2021, 29, 1361-73.
6. Rafeeq, M. M.; Murad, H. A. S. Cystic fibrosis: current therapeutic targets and future approaches. J. Transl. Med. 2017, 15, 84.
8. 2019 Chronic Respiratory Diseases Collaborators. Global burden of chronic respiratory diseases and risk factors, 1990-2019: an update from the Global Burden of Disease Study 2019. EClinicalMedicine 2023, 59, 101936.
9. Sahu, I.; Haque, A. K. M. A.; Weidensee, B.; Weinmann, P.; Kormann, M. S. D. Recent developments in mRNA-based protein supplementation therapy to target lung diseases. Mol. Ther. 2019, 27, 803-23.
10. Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J. B.; Yu, D. mRNA as a Transformative technology for vaccine development to control infectious diseases. Mol. Ther. 2019, 27, 757-72.
12. Karikó, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 2004, 279, 12542-50.
13. Cullis, P. R.; Hope, M. J. Lipid nanoparticle systems for enabling gene therapies. Mol. Ther. 2017, 25, 1467-75.
14. Chatterjee, S.; Kon, E.; Sharma, P.; Peer, D. Endosomal escape: a bottleneck for LNP-mediated therapeutics. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2307800120.
15. John, R.; Monpara, J.; Swaminathan, S.; Kalhapure, R. Chemistry and art of developing lipid nanoparticles for biologics delivery: focus on development and scale-up. Pharmaceutics 2024, 16, 131.
16. Gilbert, J.; Sebastiani, F.; Arteta, M. Y.; et al. Evolution of the structure of lipid nanoparticles for nucleic acid delivery: From in situ studies of formulation to colloidal stability. J. Colloid. Interface. Sci. 2024, 660, 66-76.
17. Mehta, M.; Bui, T. A.; Yang, X.; Aksoy, Y.; Goldys, E. M.; Deng, W. Lipid-Based nanoparticles for drug/gene delivery: an overview of the production techniques and difficulties encountered in their industrial development. ACS. Mater. Au. 2023, 3, 600-19.
18. Cheng, X.; Lee, R. J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug. Deliv. Rev. 2016, 99, 129-37.
19. Schober, G. B.; Story, S.; Arya, D. P. A careful look at lipid nanoparticle characterization: analysis of benchmark formulations for encapsulation of RNA cargo size gradient. Sci. Rep. 2024, 14, 2403.
20. Wu, S.; Lin, L.; Shi, L.; Liu, S. An overview of lipid constituents in lipid nanoparticle mRNA delivery systems. Wiley. Interdiscip. Rev. Nanomed. Nanobiotechnol. 2024, 16, e1978.
21. Zhang, Y.; Sun, C.; Wang, C.; Jankovic, K. E.; Dong, Y. Lipids and lipid derivatives for RNA delivery. Chem. Rev. 2021, 121, 12181-277.
22. Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer. 2021, 20, 41.
23. Jung, O.; Jung, H. Y.; Thuy, L. T.; et al. Modulating lipid nanoparticles with histidinamide-conjugated cholesterol for improved intracellular delivery of mRNA. Adv. Healthc. Mater. 2024, 13, e2303857.
24. Hajj, K. A.; Ball, R. L.; Deluty, S. B.; et al. Branched-tail lipid nanoparticles potently deliver mRNA in vivo due to enhanced ionization at endosomal pH. Small 2019, 15, e1805097.
25. Eygeris, Y.; Gupta, M.; Kim, J.; Sahay, G. Chemistry of lipid nanoparticles for RNA Delivery. Acc. Chem. Res. 2022, 55, 2-12.
26. Suk, J. S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L. M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug. Deliv. Rev. 2016, 99, 28-51.
27. Schoenmaker, L.; Witzigmann, D.; Kulkarni, J. A.; et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int. J. Pharm. 2021, 601, 120586.
28. Huang, X.; Ma, Y.; Ma, G.; Xia, Y. Unlocking the therapeutic applicability of LNP-mRNA: chemistry, formulation, and clinical strategies. Research. (Wash. D. C). 2024, 7, 0370.
29. Ibrahim, M.; Ramadan, E.; Elsadek, N. E.; et al. Polyethylene glycol (PEG): the nature, immunogenicity, and role in the hypersensitivity of PEGylated products. J. Control. Release. 2022, 351, 215-30.
30. Tenchov, R.; Sasso, J. M.; Zhou, Q. A. PEGylated lipid nanoparticle formulations: immunological safety and efficiency perspective. Bioconjug. Chem. 2023, 34, 941-60.
31. Zong, Y.; Lin, Y.; Wei, T.; Cheng, Q. Lipid nanoparticle (LNP) enables mRNA DElivery for cancer therapy. Adv. Mater. 2023, 35, e2303261.
32. Maeki, M.; Uno, S.; Niwa, A.; Okada, Y.; Tokeshi, M. Microfluidic technologies and devices for lipid nanoparticle-based RNA delivery. J. Control. Release. 2022, 344, 80-96.
33. Geng, C.; Zhou, K.; Yan, Y.; et al. A preparation method for mRNA-LNPs with improved properties. J. Control. Release. 2023, 364, 632-43.
34. Sackmann, E. K.; Fulton, A. L.; Beebe, D. J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181-9.
35. Damiati, S.; Kompella, U. B.; Damiati, S. A.; Kodzius, R. Microfluidic devices for drug delivery systems and drug screening. Genes. (Basel). 2018, 9, 103.
36. Shepherd, S. J.; Warzecha, C. C.; Yadavali, S.; et al. Scalable mRNA and siRNA lipid nanoparticle production using a parallelized microfluidic device. Nano. Lett. 2021, 21, 5671-80.
37. Subraveti, S. N.; Wilson, B. K.; Bizmark, N.; Liu, J.; Prud'homme, R. K. Synthesizing Lipid nanoparticles by turbulent flow in confined impinging jet mixers. J. Vis. Exp. 2024. DOI: 10.3791/67047.
38. O'Brien, L. M. N.; Costa, A. P.; Cebrero, Y. M.; et al. Process robustness in lipid nanoparticle production: a comparison of microfluidic and turbulent jet mixing. Mol. Pharm. 2023, 20, 4285-96.
39. Pratsinis, A.; Fan, Y.; Portmann, M.; et al. Impact of non-ionizable lipids and phase mixing methods on structural properties of lipid nanoparticle formulations. Int. J. Pharm. 2023, 637, 122874.
40. Cui, L.; Pereira, S.; Sonzini, S.; et al. Development of a high-throughput platform for screening lipid nanoparticles for mRNA delivery. Nanoscale 2022, 14, 1480-91.
41. Bai, C.; Wang, C.; Lu, Y. Novel vectors and administrations for mRNA delivery. Small 2023, 19, e2303713.
42. Wang, J.; Ding, Y.; Chong, K.; et al. Recent advances in lipid nanoparticles and their safety concerns for mRNA delivery. Vaccines. (Basel). 2024, 12, 1148.
43. Wang, C.; Zhang, Y.; Dong, Y. Lipid nanoparticle-mRNA formulations for therapeutic applications. Acc. Chem. Res. 2021, 54, 4283-93.
44. Xiao, Y.; Tang, Z.; Huang, X.; et al. Emerging mRNA technologies: delivery strategies and biomedical applications. Chem. Soc. Rev. 2022, 51, 3828-45.
45. El-Mayta, R.; Padilla, M. S.; Billingsley, M. M.; Han, X.; Mitchell, M. J. Testing the in vitro and in vivo efficiency of mRNA-lipid nanoparticles formulated by microfluidic mixing. J. Vis. Exp. 2023. DOI: 10.3791/64810.
46. Liu, Y.; Huang, Y.; He, G.; Guo, C.; Dong, J.; Wu, L. Development of mRNA lipid nanoparticles: targeting and therapeutic aspects. Int. J. Mol. Sci. 2024, 25, 10166.
47. Hajiaghapour, A. M.; Dayani, F.; Saedi, S. F.; et al. Lipid nanoparticles as promising carriers for mRNA vaccines for viral lung infections. Pharmaceutics 2023, 15, 1127.
48. Cullis, P. R.; Felgner, P. L. The 60-year evolution of lipid nanoparticles for nucleic acid delivery. Nat. Rev. Drug. Discov. 2024, 23, 709-22.
49. Igyártó, B. Z.; Qin, Z. The mRNA-LNP vaccines - the good, the bad and the ugly? Front. Immunol. 2024, 15, 1336906.
50. Liu, J.; Cabral, H.; Mi, P. Nanocarriers address intracellular barriers for efficient drug delivery, overcoming drug resistance, subcellular targeting and controlled release. Adv. Drug. Deliv. Rev. 2024, 207, 115239.
51. Jiao, X.; He, X.; Qin, S.; et al. Insights into the formulation of lipid nanoparticles for the optimization of mRNA therapeutics. Wiley. Interdiscip. Rev. Nanomed. Nanobiotechnol. 2024, 16, e1992.
52. Weng, Y.; Xiao, H.; Zhang, J.; Liang, X. J.; Huang, Y. RNAi therapeutic and its innovative biotechnological evolution. Biotechnol. Adv. 2019, 37, 801-25.
53. Barbier, A. J.; Jiang, A. Y.; Zhang, P.; Wooster, R.; Anderson, D. G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022, 40, 840-54.
54. Shaw, C. A.; Essink, B.; Harper, C.; et al. Safety and immunogenicity of an mRNA-based RSV vaccine including a 12-month booster in a phase 1 clinical trial in healthy older adults. J. Infect. Dis. 2024, 230, e647-56.
55. Hanada, S.; Pirzadeh, M.; Carver, K. Y.; Deng, J. C. Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front. Immunol. 2018, 9, 2640.
56. Maginnis, M. S. Virus-receptor interactions: the key to cellular invasion. J. Mol. Biol. 2018, 430, 2590-611.
57. Herold, S.; Becker, C.; Ridge, K. M.; Budinger, G. R. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur. Respir. J. 2015, 45, 1463-78.
58. Arevalo, C. P.; Bolton, M. J.; Le, S. V.; et al. A multivalent nucleoside-modified mRNA vaccine against all known influenza virus subtypes. Science 2022, 378, 899-904.
59. Bahl, K.; Senn, J. J.; Yuzhakov, O.; et al. Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol. Ther. 2017, 25, 1316-27.
60. Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078-94.
61. Magini, D.; Giovani, C.; Mangiavacchi, S.; et al. Self-amplifying mRNA vaccines expressing multiple conserved influenza antigens confer protection against homologous and heterosubtypic viral challenge. PLoS. One. 2016, 11, e0161193.
62. Lokugamage, M. P.; Vanover, D.; Beyersdorf, J.; et al. Optimization of lipid nanoparticles for the delivery of nebulized therapeutic mRNA to the lungs. Nat. Biomed. Eng. 2021, 5, 1059-68.
63. Zhuang, X.; Chen, L.; Yang, S.; et al. R848 Adjuvant laden with self-assembled nanoparticle-based mRNA vaccine elicits protective immunity against H5N1 in mice. Front. Immunol. 2022, 13, 836274.
64. Jackson, C. B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell. Biol. 2022, 23, 3-20.
65. Gusev, E.; Sarapultsev, A.; Solomatina, L.; Chereshnev, V. SARS-CoV-2-specific immune response and the pathogenesis of COVID-19. Int. J. Mol. Sci. 2022, 23, 1716.
66. Meo, S. A.; Bukhari, I. A.; Akram, J.; Meo, A. S.; Klonoff, D. C. COVID-19 vaccines: comparison of biological, pharmacological characteristics and adverse effects of Pfizer/BioNTech and moderna vaccines. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1663-9.
67. Khurana, A.; Allawadhi, P.; Khurana, I.; et al. Role of nanotechnology behind the success of mRNA vaccines for COVID-19. Nano. Today. 2021, 38, 101142.
68. Chen, K.; Fan, N.; Huang, H.; et al. mRNA vaccines against SARS-CoV-2 variants delivered by lipid nanoparticles based on novel ionizable lipids. Adv. Funct. Mater. 2022, 32, 2204692.
69. Venit, T.; Blavier, J.; Maseko, S. B.; et al. Nanobody against SARS-CoV-2 non-structural protein Nsp9 inhibits viral replication in human airway epithelia. Mol. Ther. Nucleic. Acids. 2024, 35, 102304.
70. Ma, Q.; Li, R.; Guo, J.; et al. Immunization with a prefusion SARS-CoV-2 spike protein vaccine (RBMRNA-176) protects against viral challenge in mice and nonhuman primates. Vaccines. (Basel). 2022, 10, 1698.
71. Tai, W.; Yang, K.; Liu, Y.; et al. A lung-selective delivery of mRNA encoding broadly neutralizing antibody against SARS-CoV-2 infection. Nat. Commun. 2023, 14, 8042.
72. Uraki, R.; Imai, M.; Ito, M.; et al. An mRNA vaccine encoding the SARS-CoV-2 receptor-binding domain protects mice from various Omicron variants. NPJ. Vaccines. 2024, 9, 4.
73. Li, J.; Xiao, L.; Chen, Z.; et al. A spike-based mRNA vaccine that induces durable and broad protection against porcine deltacoronavirus in piglets. J. Virol. 2024, 98, e0053524.
74. Kim, J.; Jozic, A.; Mukherjee, A.; et al. Rapid generation of circulating and mucosal decoy human ACE2 using mRNA nanotherapeutics for the potential treatment of SARS-CoV-2. Adv. Sci. (Weinh). 2022, 9, e2202556.
75. Milligan, E. C.; Olstad, K.; Williams, C. A.; et al. Infant rhesus macaques immunized against SARS-CoV-2 are protected against heterologous virus challenge 1 year later. Sci. Transl. Med. 2023, 15, eadd6383.
76. Zhao, H.; Wang, T. C.; Li, X. F.; et al. Long-term stability and protection efficacy of the RBD-targeting COVID-19 mRNA vaccine in nonhuman primates. Signal. Transduct. Target. Ther. 2021, 6, 438.
77. Qin, J.; Jeon, J. H.; Xu, J.; et al. Design and preclinical evaluation of a universal SARS-CoV-2 mRNA vaccine. Front. Immunol. 2023, 14, 1126392.
78. Ye, Z.; Bonam, S. R.; McKay, L. G. A.; et al. Monovalent SARS-COV-2 mRNA vaccine using optimal UTRs and LNPs is highly immunogenic and broadly protective against Omicron variants. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2311752120.
79. Polack, F. P.; Thomas, S. J.; Kitchin, N.; et al. C4591001 Clinical Trial Group. Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. N. Engl. J. Med. 2020, 383, 2603-15.
80. Baden, L. R.; El, S. H. M.; Essink, B.; et al. COVE Study Group. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021, 384, 403-16.
81. Dagan, N.; Barda, N.; Kepten, E.; et al. BNT162b2 mRNA covid-19 vaccine in a nationwide mass vaccination setting. N. Engl. J. Med. 2021, 384, 1412-23.
82. Corbett, K. S.; Edwards, D. K.; Leist, S. R.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567-71.
83. Borchers, A. T.; Chang, C.; Gershwin, M. E.; Gershwin, L. J. Respiratory syncytial virus - a comprehensive review. Clin. Rev. Allergy. Immunol. 2013, 45, 331-79.
84. Garcia-Garcia, M. L.; Calvo, R. C.; Del, R. R. T. Pediatric asthma and viral infection. Arch. Bronconeumol. 2016, 52, 269-73.
85. Qiu, X.; Xu, S.; Lu, Y.; et al. Development of mRNA vaccines against respiratory syncytial virus (RSV). Cytokine. Growth. Factor. Rev. 2022, 68, 37-53.
86. Wu, N.; Zhang, J.; Shen, Y.; et al. A potential bivalent mRNA vaccine candidate protects against both RSV and SARS-CoV-2 infections. Mol. Ther. 2024, 32, 1033-47.
88. Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis updated. Circ. Res. 2020, 127, 310-29.
89. Popoola, D. O.; Cao, Z.; Men, Y.; et al. Lung-specific mRNA delivery enabled by sulfonium lipid nanoparticles. Nano. Lett. 2024, 24, 8080-8.
90. Liu, G. Y.; Budinger, G. R. S.; Dematte, J. E. Advances in the management of idiopathic pulmonary fibrosis and progressive pulmonary fibrosis. BMJ. 2022, 377, e066354.
91. Martinez, F. J.; Collard, H. R.; Pardo, A.; et al. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers. 2017, 3, 17074.
92. Spagnolo, P.; Kropski, J. A.; Jones, M. G.; et al. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol. Ther. 2021, 222, 107798.
93. Moss, B. J.; Ryter, S. W.; Rosas, I. O. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2022, 17, 515-46.
94. Richeldi, L.; Collard, H. R.; Jones, M. G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941-52.
95. Massaro, M.; Wu, S.; Baudo, G.; et al. Lipid nanoparticle-mediated mRNA delivery in lung fibrosis. Eur. J. Pharm. Sci. 2023, 183, 106370.
96. Wang, Y.; Zhang, J.; Liu, Y.; et al. Realveolarization with inhalable mucus-penetrating lipid nanoparticles for the treatment of pulmonary fibrosis in mice. Sci. Adv. 2024, 10, eado4791.
97. Zhang, R.; Jing, W.; Chen, C.; et al. Inhaled mRNA Nanoformulation with biogenic ribosomal protein reverses established pulmonary fibrosis in a bleomycin-induced murine model. Adv. Mater. 2022, 34, e2107506.
98. Rajendran, P.; Rengarajan, T.; Thangavel, J.; et al. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057-69.
99. Augustin, H. G.; Koh, G. Y. Organotypic vasculature: from descriptive heterogeneity to functional pathophysiology. Science 2017, 357, eaal2379.
100. Teijaro, J. R.; Walsh, K. B.; Cahalan, S.; et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011, 146, 980-91.
101. Radloff, K.; Gutbier, B.; Dunne, C. M.; et al. Cationic LNP-formulated mRNA expressing Tie2-agonist in the lung endothelium prevents pulmonary vascular leakage. Mol. Ther. Nucleic. Acids. 2023, 34, 102068.
102. Zhao, G.; Xue, L.; Weiner, A. I.; et al. TGF-βR2 signaling coordinates pulmonary vascular repair after viral injury in mice and human tissue. Sci. Transl. Med. 2024, 16, eadg6229.
103. McCarthy, C.; Gupta, N.; Johnson, S. R.; Yu, J. J.; McCormack, F. X. Lymphangioleiomyomatosis: pathogenesis, clinical features, diagnosis, and management. Lancet. Respir. Med. 2021, 9, 1313-27.
104. Barrera E, Mancheño Franch N, Vera-Sempere F, Padilla Alarcón J. Lymphangioleiomyomatosis. Arch. Bronconeumol. 2011, 47, 85-93.
105. Kundu, N.; Holz, M. K. Lymphangioleiomyomatosis: a metastatic lung disease. Am. J. Physiol. Cell. Physiol. 2023, 324, C320-6.
106. Bhaoighill, M. N.; Dunlop, E. A. Mechanistic target of rapamycin inhibitors: successes and challenges as cancer therapeutics. Cancer. Drug. Resist. 2019, 2, 1069-85.
107. Qiu, M.; Tang, Y.; Chen, J.; et al. Lung-selective mRNA delivery of synthetic lipid nanoparticles for the treatment of pulmonary lymphangioleiomyomatosis. Proc. Natl. Acad. Sci. U. S. A. 2022, 119.
108. Remark, R.; Becker, C.; Gomez, J. E.; et al. The non-small cell lung cancer immune contexture. A major determinant of tumor characteristics and patient outcome. Am. J. Respir. Crit. Care. Med. 2015, 191, 377-90.
109. Jones, G. S.; Baldwin, D. R. Recent advances in the management of lung cancer. Clin. Med. (Lond). 2018, 18, s41-6.
110. Feng, J.; Zhang, P.; Wang, D.; Li, Y.; Tan, J. New strategies for lung cancer diagnosis and treatment: applications and advances in nanotechnology. Biomark. Res. 2024, 12, 136.
111. Cheng, X.; Liu, Q.; Li, H.; et al. Lipid nanoparticles loaded with an antisense oligonucleotide gapmer against Bcl-2 for treatment of lung cancer. Pharm. Res. 2017, 34, 310-20.
112. Cheng, X.; Yu, D.; Cheng, G.; et al. T7 Peptide-conjugated lipid nanoparticles for dual modulation of Bcl-2 and Akt-1 in lung and cervical carcinomas. Mol. Pharm. 2018, 15, 4722-32.
113. Yu, J.; Li, Q.; Zhang, C.; et al. Targeted LNPs deliver IL-15 superagonists mRNA for precision cancer therapy. Biomaterials 2025, 317, 123047.
114. Hussain, M. S.; Sultana, A.; Bisht, A. S.; Gupta, G. Groundbreaking mRNA lung cancer vaccine trials: a new dawn in cancer treatment. Curr. Cancer. Drug. Targets. , 2025, 1082-7.
115. Kon, E.; Ad-El, N.; Hazan-Halevy, I.; Stotsky-Oterin, L.; Peer, D. Targeting cancer with mRNA-lipid nanoparticles: key considerations and future prospects. Nat. Rev. Clin. Oncol. 2023, 20, 739-54.
116. Kiaie, S. H.; Majidi, Z. N.; Ahmadi, A.; et al. Recent advances in mRNA-LNP therapeutics: immunological and pharmacological aspects. J. Nanobiotechnology. 2022, 20, 276.
117. Rospond, B.; Krakowska, A.; Muszyńska, B.; Opoka, W. The history, current state and perspectives of aerosol therapy. Acta. Pharm. 2022, 72, 225-43.
118. van, R. C. J. M.; Vlaming, K. E.; Bem, R. A.; et al. Low energy nebulization preserves integrity of SARS-CoV-2 mRNA vaccines for respiratory delivery. Sci. Rep. 2023, 13, 8851.
119. Yan, R.; Zou, C.; Yang, X.; et al. Nebulized inhalation drug delivery: clinical applications and advancements in research. J. Mater. Chem. B. 2025, 13, 821-43.
120. Miao, H.; Huang, K.; Li, Y.; et al. Optimization of formulation and atomization of lipid nanoparticles for the inhalation of mRNA. Int. J. Pharm. 2023, 640, 123050.
121. Kim, J.; Jozic, A.; Lin, Y.; et al. Engineering Lipid Nanoparticles for Enhanced Intracellular Delivery of mRNA through Inhalation. ACS. Nano. 2022, 16, 14792-806.
122. Jiang, A. Y.; Witten, J.; Raji, I. O.; et al. Combinatorial development of nebulized mRNA delivery formulations for the lungs. Nat. Nanotechnol. 2024, 19, 364-75.
123. Bai, X.; Chen, Q.; Li, F.; et al. Optimized inhaled LNP formulation for enhanced treatment of idiopathic pulmonary fibrosis via mRNA-mediated antibody therapy. Nat. Commun. 2024, 15, 6844.
124. Liu, S.; Wen, Y.; Shan, X.; et al. Charge-assisted stabilization of lipid nanoparticles enables inhaled mRNA delivery for mucosal vaccination. Nat. Commun. 2024, 15, 9471.
125. Courrier, H. M.; Butz, N.; Vandamme, T. F. Pulmonary drug delivery systems: recent developments and prospects. Crit. Rev. Ther. Drug. Carrier. Syst. 2002, 19, 425-98.
126. Molina, R. M.; Konduru, N. V.; Hirano, H.; et al. Pulmonary distribution of nanoceria: comparison of intratracheal, microspray instillation and dry powder insufflation. Inhal. Toxicol. 2016, 28, 550-60.
127. Wang, H.; Wu, L.; Sun, X. Intratracheal delivery of nano- and microparticles and hyperpolarized gases: a promising strategy for the imaging and treatment of respiratory disease. Chest 2020, 157, 1579-90.
128. Tafech, B.; Rokhforouz, M. R.; Leung, J.; et al. Exploring mechanisms of lipid nanoparticle-mucus interactions in healthy and cystic fibrosis conditions. Adv. Healthc. Mater. 2024, 13, e2304525.
129. Zhang, H.; Leal, J.; Soto, M. R.; Smyth, H. D. C.; Ghosh, D. Aerosolizable lipid nanoparticles for pulmonary delivery of mrna through design of experiments. Pharmaceutics 2020, 12, 1042.
130. Friis, K. P.; Gracin, S.; Oag, S.; et al. Spray dried lipid nanoparticle formulations enable intratracheal delivery of mRNA. J. Control. Release. 2023, 363, 389-401.
131. Sarode, A.; Patel, P.; Vargas-Montoya, N.; et al. Inhalable dry powder product (DPP) of mRNA lipid nanoparticles (LNPs) for pulmonary delivery. Drug. Deliv. Transl. Res. 2024, 14, 360-72.
132. Beck, S. E.; Laube, B. L.; Barberena, C. I.; et al. Deposition and expression of aerosolized rAAV vectors in the lungs of Rhesus macaques. Mol. Ther. 2002, 6, 546-54.
133. Pardi, N.; Tuyishime, S.; Muramatsu, H.; et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release. 2015, 217, 345-51.
134. Geng, L.; Kato, N.; Kodama, Y.; Mukai, H.; Kawakami, S. Influence of lipid composition of messenger RNA-loaded lipid nanoparticles on the protein expression via intratracheal administration in mice. Int. J. Pharm. 2023, 637, 122896.
135. Tam, A.; Kulkarni, J.; An, K.; et al. Lipid nanoparticle formulations for optimal RNA-based topical delivery to murine airways. Eur. J. Pharm. Sci. 2022, 176, 106234.
137. Zhang, W.; Chetwynd, A. J.; Thorn, J. A.; Lynch, I.; Ramautar, R. Understanding the significance of sample preparation in studies of the nanoparticle metabolite corona. ACS. Meas. Sci. Au. 2022, 2, 251-60.
138. Ke, P. C.; Lin, S.; Parak, W. J.; Davis, T. P.; Caruso, F. A decade of the protein corona. ACS. Nano. 2017, 11, 11773-6.
139. Walczyk, D.; Bombelli, F. B.; Monopoli, M. P.; Lynch, I.; Dawson, K. A. What the cell “sees” in bionanoscience. J. Am. Chem. Soc. 2010, 132, 5761-8.
140. Lynch, I.; Dawson, K. A.; Linse, S. Detecting cryptic epitopes created by nanoparticles. Sci. STKE. 2006, 2006, pe14.
141. Monopoli, M. P.; Aberg, C.; Salvati, A.; Dawson, K. A. Biomolecular coronas provide the biological identity of nanosized materials. Nat. Nanotechnol. 2012, 7, 779-86.
142. Petersen, D. M. S.; Weiss, R. M.; Hajj, K. A.; et al. Branched-tail lipid nanoparticles for intravenous mRNA delivery to lung immune, endothelial, and alveolar cells in mice. Adv. Healthc. Mater. 2024, 13, e2400225.
143. Eygeris, Y.; Gupta, M.; Kim, J.; et al. Thiophene-based lipids for mRNA delivery to pulmonary and retinal tissues. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2307813120.
144. Kim, M.; Jeong, M.; Lee, G.; et al. Novel piperazine-based ionizable lipid nanoparticles allow the repeated dose of mRNA to fibrotic lungs with improved potency and safety. Bioeng. Transl. Med. 2023, 8, e10556.
145. Kowalski, P. S.; Capasso, P. U.; Huang, Y.; Rudra, A.; Langer, R.; Anderson, D. G. Ionizable amino-polyesters synthesized via ring opening polymerization of tertiary amino-alcohols for tissue selective mRNA delivery. Adv. Mater. 2018, e1801151.
146. Guéguen, C.; Ben, C. T.; Briand, M.; et al. Evaluating how cationic lipid affects mRNA-LNP physical properties and biodistribution. Eur. J. Pharm. Biopharm. 2024, 195, 114077.
147. Zeng, G.; He, Z.; Yang, H.; et al. Cationic lipid pairs enhance liver-to-lung tropism of lipid nanoparticles for in vivo mRNA delivery. ACS. Appl. Mater. Interfaces. 2024, 16, 25698-709.
148. Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L. T.; Dilliard, S. A.; Siegwart, D. J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313-20.
149. Wei, T.; Sun, Y.; Cheng, Q.; et al. Lung SORT LNPs enable precise homology-directed repair mediated CRISPR/Cas genome correction in cystic fibrosis models. Nat. Commun. 2023, 14, 7322.
150. Dabke, A.; Ghosh, S.; Dabke, P.; Sawant, K.; Khopade, A. Revisiting the in-vitro and in-vivo considerations for in-silico modelling of complex injectable drug products. J. Control. Release. 2023, 360, 185-211.
151. Sun, Y.; Chatterjee, S.; Lian, X.; et al. In vivo editing of lung stem cells for durable gene correction in mice. Science 2024, 384, 1196-202.
152. Dilliard, S. A.; Cheng, Q.; Siegwart, D. J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2021, 118.
153. Vayalakkara, R. K.; Lo, C. L.; Chen, H. H.; et al. Photothermal/NO combination therapy from plasmonic hybrid nanotherapeutics against breast cancer. J. Control. Release. 2022, 345, 417-32.
154. Radmand, A.; Kim, H.; Beyersdorf, J.; et al. Cationic cholesterol-dependent LNP delivery to lung stem cells, the liver, and heart. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2307801120.
155. Fei, Y.; Yu, X.; Liu, P.; Ren, H.; Wei, T.; Cheng, Q. Simplified lipid nanoparticles for tissue- and cell-targeted mRNA delivery facilitate precision tumor therapy in a lung metastasis mouse model. Adv. Mater. 2024, 36, e2409812.
156. Tkachenko, E.; Tse, D.; Sideleva, O.; et al. Caveolae, fenestrae and transendothelial channels retain PV1 on the surface of endothelial cells. PLoS. One. 2012, 7, e32655.
157. Parton, R. G.; del, P. M. A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell. Biol. 2013, 14, 98-112.
Cite This Article

How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].