



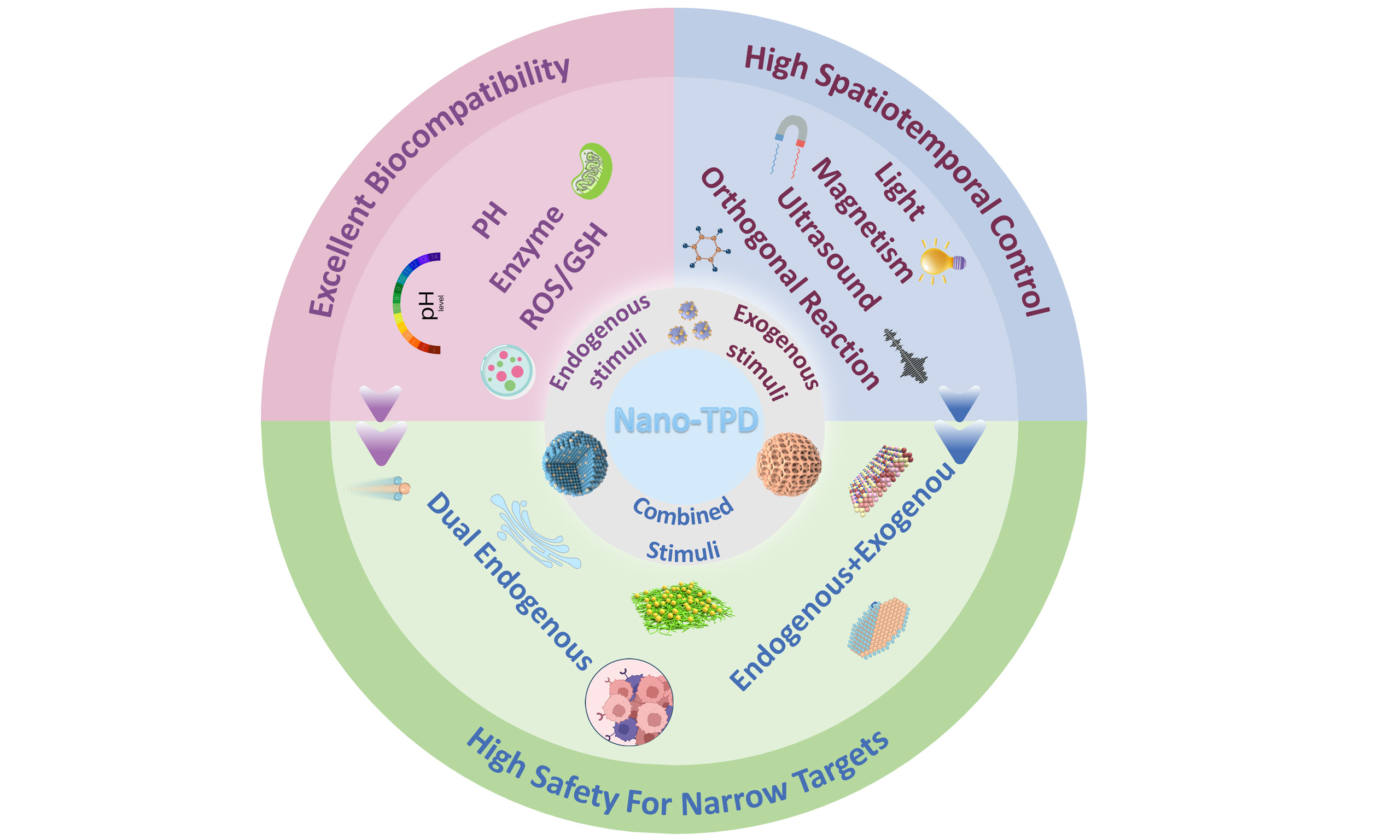
Nano-TPD for precision protein degradation via endogenous or exogenous triggered release
 0
0 Abstract
Targeted protein degradation (TPD) technology offers a revolutionary approach for precise intervention against “undruggable” targets by exploiting intracellular pathways such as the ubiquitin-proteasome system and the autophagy-lysosome system. However, its clinical translation faces significant challenges, including limited delivery efficiency, poor tissue specificity, and off-target toxicity. The integration of nanotechnology with TPD not only overcomes the physicochemical limitations of traditional TPD molecules [e.g., Proteolysis-Targeting Chimeras (PROTACs), Autophagosome-Tethering Compounds (ATTECs), Lysosome-Targeting Chimeras (LYTACs), molecular glues] through the design of functionalized carriers but also equips these systems with spatiotemporal precision activation via diverse regulation of “initiation conditions”. This review systematically traces the developmental trajectory and fundamental mechanisms of TPD technology, explores the delivery characteristics and principles of functional modification in nanocarriers, and comprehensively discusses the design strategies, mechanisms, and recent advances of nanotyped TPD systems categorized by their initiation conditions: endogenous microenvironment-responsive [pH, enzyme, reactive oxygen species (ROS)/glutathione (GSH)], exogenous stimulus-initiated (light, magnetic, ultrasound, bioorthogonal reaction), and multi-conditionally co-initiated systems (endogenous-endogenous and endogenous-exogenous signal synergy). Furthermore, this review highlights key challenges in the field, such as carrier biocompatibility, modulation of degradation efficiency, and in vivo fate tracking, while outlining critical directions for clinical translation, including personalized customization, intelligent logic gating, and multimodal characterization. By providing a systematic theoretical framework and practical insights, this review aims to promote cross-disciplinary integration and foster innovative drug development at the intersection of nanotechnology and TPD.
Keywords
INTRODUCTION
Traditional drug development strategies have primarily focused on inhibiting or activating the functions of target proteins[1]. However, many diseases arise not from abnormal protein function, but from the aberrant accumulation or overexpression of proteins within cells[2]. These proteins often carry significant clinical relevance yet remain challenging to target effectively. To overcome these limitations, strategies that directly eliminate abnormal proteins have become increasingly vital[3]. In recent years, Targeted Protein Degradation (TPD) technology has emerged as a promising solution. This encompasses approaches such as Proteolysis-Targeting Chimeras (PROTACs), Molecular Glue Degraders, Autophagy-Targeting Chimeras (AUTACs), Autophagosome-Tethering Compounds (ATTECs), Lysosome-Targeting Chimeras (LYTACs), and related derivatives[4]. Operating through an “event-driven” catalytic mode, TPD functions effectively at sub-stoichiometric ratios, overcoming the limitations of traditional small-molecule drugs by precisely targeting previously intractable proteins[5-9]. This breakthrough opens new avenues for drug development. As an innovative therapeutic strategy, the core advantage of TPD lies in its ability to induce the degradation of specific intracellular proteins rather than merely inhibiting enzymatic activity[10]. By harnessing the cell’s intrinsic Ubiquitin-Proteasome System (UPS) or lysosomal degradation pathways, TPD has demonstrated substantial potential across various disease areas, including cancer and neurodegenerative disorders[11].
Nevertheless, each TPD technology has inherent limitations, and clinical translation remains constrained by multiple intrinsic challenges[12]. For example, PROTACs typically possess large molecular weights (> 800 Da), violating Lipinski’s Rule of Five, which leads to poor membrane permeability, low oral bioavailability, off-target toxicity, and the potential for a “hook effect”[13-16]. Although Molecular Glue Degraders benefit from favorable molecular size and drug-likeness, their mode of action depends on inducing novel protein - protein interaction interfaces, making molecular discovery heavily reliant on empirical screening and posing significant obstacles to rational design[17]. Moreover, molecules containing electrophilic groups may cause non-specific protein modifications and broad-spectrum cytotoxicity[18]. In autophagy-based strategies like ATTECs, degradation efficiency is highly dependent on endogenous autophagic flux, which varies considerably among tissues and disease states, complicating efforts to achieve stable and tumor-selective degradation[19]. Lysosome-targeting technologies such as LYTACs have broadened the target scope to include membrane and secreted proteins; however, they face challenges related to complex molecular structures, limited tissue penetration, and non-specific receptor expression that can induce degradation in normal tissues[20,21]. Thus, further optimization is needed to enhance monotherapy efficacy and safety. Collectively, these challenges suggest that focusing solely on structural optimization of TPD molecules is insufficient to overcome the bottlenecks hindering clinical translation[22].
Nanocarriers, as advanced drug delivery platforms, offer a highly promising strategy to address the limitations of TPD technology[23]. Typically ranging in size from 1 to 1,000 nanometers, nanocarriers, including liposomes, polymeric nanoparticles, and micelles, can encapsulate and protect drug molecules, prolong their circulation time in the bloodstream, and preferentially accumulate at target sites via the enhanced permeability and retention (EPR) effect or through active targeting using ligands such as antibodies or peptides[24]. Moreover, nanocarriers can be engineered to respond to tumor microenvironmental cues, such as acidic pH, elevated glutathione levels, and reactive oxygen species, or to external stimuli like light and heat, enabling precise spatiotemporal control over drug release. These capabilities have significantly advanced the pharmacokinetics and therapeutic efficacy of a wide range of therapeutics, including small molecules, nucleic acids, and protein-based drugs[25]. Furthermore, the surface engineering of nanocarriers can optimize the interfacial interactions between biomolecules and functional substrates, demonstrating that nanotechnology exhibits broad applicability in enhancing the performance of bioactive molecules, extending beyond traditional drug delivery[26].
The integration of nanotechnology with TPD strategies has demonstrated significant advantages, effectively addressing the limitations of various TPD approaches[27]. For instance, nano-PROTACs enhance cellular permeability and bioavailability by encapsulating PROTACs at the nanoscale, promoting accumulation in tumor tissues via the EPR effect, and minimizing off-target toxicity through surface functionalization or stimulus-responsive release[28]. Self-assembled, stimulus-responsive nano-molecular glues, designed to respond to elevated glutathione (GSH) or hydrogen peroxide (H2O2) levels in the tumor microenvironment (TME), enable tumor-specific release, achieving targeted degradation while effectively mitigating broad-spectrum cytotoxicity[29]. Nano-ATTEC systems, based on mixed-shell polymeric micelles, respond to the acidic TME, facilitating tumor-selective uptake and simultaneously activating autophagy to establish a positive feedback loop that enhances degradation efficiency[30]. Moreover, the bifunctional LYTAC nanoplatform (NLTC) achieves efficient membrane protein degradation through surface modification with lysosome-targeting and target protein ligands. By encapsulating functional enzymes such as catalase and incorporating pH-responsive, cleavable polyethylene glycol (PEG) shells, NLTC synergistically modulates the tumor immune microenvironment, enabling tumor-selective degradation alongside enhanced immunotherapeutic effects[31].
The combination of nanotechnology with diverse TPD strategies, including PROTACs, Molecular Glues, ATTECs, LYTACs, and so on, offers a powerful engineering platform for the precise and selective elimination of “undruggable” targets, particularly tumor-associated pathogenic proteins[32]. Nanocarriers not only address the inherent limitations of TPD molecules in pharmacokinetics, biodistribution, and safety but also provide a foundation for developing “programmable degradation systems” featuring multi-signal integration and intelligent on-off control[33]. In this context, the design and optimization of “activation triggers” for nano-TPD systems, including endogenous stimuli [pH, enzymes, or reactive oxygen species (ROS)/GSH], exogenous triggers (light, ultrasound, magnetic fields, bioorthogonal reactions), and combined dual-trigger modalities, have become critical scientific challenges for achieving high selectivity, safety, and efficacy[11]. Building upon a systematic review of TPD technology’s developmental landscape and core mechanisms, as well as an in-depth analysis of nanocarrier delivery characteristics and functional modification principles, this review centers on the “activation triggers” of nano-TPD systems as a primary classification criterion [Table 1]. It comprehensively discusses the design strategies, mechanisms of action, and recent advances in endogenous microenvironment-responsive, exogenous stimulus-initiated, and multi-condition combined activation systems. Furthermore, it summarizes key challenges, including carrier biocompatibility, regulation of degradation efficiency, and in vivo fate tracking, and highlights promising directions for clinical translation such as personalized customization, intelligent logic gating, and multimodal characterization. This work aims to provide a systematic theoretical framework and practical guidance to promote interdisciplinary integration of nanotechnology and TPD, ultimately advancing innovative drug development.
The representative nano-TPD and its mechanism of action
| Name | Types of nanocarriers | TPD type | Trigger condition | Target protein | Ref. |
| MNC@Ca/MnCO3 /ARV/anti-PD1 | Nanocomposites | PROTAC | pH | BRD4 | [46] |
| MSPMs | Polymeric micelles | ATTEC | pH | BRD4/AR | [30] |
| NLTC | Protein nanocapsules | LYTAC | pH | Tumor-selective protein | [31] |
| EGFR/PDL1 IMTACs | DNA nanocircles | LYTAC | pH | EGFR/PDL1 | [49] |
| i16-ETNc | Amphiphilic conjugates | PROTAC | pH | LRG1 | [50] |
| Supra-LYTAC | Amphiphilic peptides | LYTAC | Enzyme | POI | [54] |
| dBET6@CFMPD | Conjugates | PROTAC | Enzyme | BRD4 | [56] |
| LipoSM- PROTAC | Liposomes | PROTAC | Enzyme | ERα | [57] |
| SM-PROTAC | Nanospheres | PROTAC | Enzyme | ERα/CDK4/6/BRD2/4 | [58] |
| Pre-PROTACs | Small-molecule conjugates | PROTAC | ROS | BRD3 | [65] |
| BT-NanoPROTAC | Small-molecule conjugates | PROTAC | ROS | BRD4 | [66] |
| TCO-MZ1 | Nanofiber | PROTAC | ROS | PD-L1 | [67] |
| ARV@PDSA | Polymers | PROTAC | GSH | HER2 | [68] |
| Cle-NP | Amphiphilicn nanoparticles | Molecular glue | GSH | Bcr-Abl | [29] |
| NanoCLY | Amphiphilic chimeras | LYTAC | GSH | CTGF | [69] |
| USDPR | Cationic polymers | PROTAC | Light | BRD4 | [86] |
| UMSNs@phoBET1 | Nanocage | PROTAC | Light | BRD4 | [87] |
| NAP | Conjugates | PROTAC | Light | BRD4 | [89] |
| SPNly | Polymeric chimers | LYTAC | Ultrasound | IL-4R | [94] |
| MCM/ARV | Nanoclusters | PROTAC | Magnetic | BRD4 | [104] |
| MZ1-O@NP | Nanoparticles | PROTAC | Click reaction | BRD4 | [108] |
| Nano-CLIPTACs | Liposomes | PROTAC | Click reaction | ALK | [111] |
| POLY-PROTAC | Amphiphilic polymers | PROTAC | Click reaction | BRD4 | [112] |
| ENCTACs | Small-molecule fragment | PROTAC | Enzymatic orthogonal reaction | BRD4 | [113] |
| PSRNs | Block copolymers | PROTAC | pH & enzyme | CDK4/6 | [124] |
| RLDPB | Micelles | PROTAC | pH & GSH | BRD4 | [125] |
| CREATE | Polymers | PROTAC | pH & GSH | BRD4 | [126] |
| CuS-MD@CS | Inorganic nanoparticles | PROTAC | ROS & Copper Ion | MDM2 | [127] |
| NanoTAC | Conjugates | PROTAC | Light & enzyme | HK2 | [130] |
| LPN | Conjugates | PROTAC | Light & enzyme | IDO | [131] |
| SPNpro | Semiconducting polymers | PROTAC | Light & enzyme | IDO | [132] |
| AZO-PRO | Conjugates | PROTAC | Light & hypoxia | BRD4 | [133] |
| ARV@PEG-ICG | Polymeric nanoparticles | PROTAC | Light & pH | BRD4 | [134] |
| HSA@PPa-JQ1 | Biomacromolecular | PDTAC | Light & pH & GSH | BRD4 | [135] |
| PGDAT@N | Polymers | PROTAC | Light & enzyme & hypoxia & pH | BRD4 | [136] |
| NP(Ce6+PRO) | Polymers | PROTAC | Ultrasonic & GSH | BRD4 | [137] |
| EMPLANT | Polymers | PROTAC | Magnetic & pH | TRIM24 | [138] |
ENDOGENOUS CONDITION INITIATION OF NANO-TPD SYSTEMS
By leveraging endogenous pathological signals as activation triggers, this approach enables protein degradation through microenvironment-responsive mechanisms. Nanoparticles are designed to respond selectively to pathological environments while remaining stable in healthy tissues. Upon entry into diseased regions, elevated biological signals induce structural changes in nanoparticles, thereby initiating degradation [Table 2]. A key advantage is intelligent microenvironment adaptation without external intervention[34]. However, variability in endogenous signals may result in inconsistent activation efficiency[35].
Conditionally starting nano-targeted degradation system
| Response conditions | Trigger mechanism | Degradation pathway | Targeting strategy | Efficiency-influencing factors | Specificity guarantee mechanism | Ref. |
| Endogenously initiated | ||||||
| pH | Acidic microenvironment → Protonation → Carrier dissociation/Degrader exposure | UPS/Lysosomal pathway | EPR (Enhanced Permeability and Retention) effect & Active targeting & Endosomal escape assistance | Environmental pH value, protonation rate, carrier hydrophilic-hydrophobic balance | Acidic microenvironment-specific triggering & Ligand-receptor targeted binding | [30,31,46,49,50] |
| Enzyme | Tumor-overexpressed enzymes → Enzymatic cleavage → Carrier depolymerization/PROTAC activation | UPS | Enzyme expression targeting & Active targeting | Enzyme expression level, enzymatic hydrolysis efficiency, peptide bond specificity | Enzyme-substrate specific recognition & Tumor microenvironment enzyme overexpression enrichment | [54,56-58] |
| ROS | Tumor high ROS level → Oxidative cleavage → PROTAC release | UPS | EPR effect & ROS-enriched targeting & Active targeting | ROS concentration, oxidation rate of sensitive bonds, carrier stability | Tumor high ROS microenvironment specificity & Sensitive bond oxidative selectivity | [65-67] |
| GSH | Intracellular high GSH concentration → Disulfide bond cleavage → Carrier dissociation | UPS | Intracellular GSH targeting & Active targeting | GSH concentration gradient, number of disulfide bonds, reduction rate | Specificity of intracellular-extracellular GSH concentration difference & Active targeting-mediated intracellular delivery | [29,68,69] |
| Exogenously initiated | ||||||
| Light | Photoisomerization/Photolytic cleavage → PROTAC core exposure/Nanocarrier dissociation | UPS | EPR effect & Light-targeted enrichment & Active targeting-mediated intracellular delivery | Light irradiation power, trigger time, PROTAC release rate | Spatiotemporally precise light irradiation control & Ligand-receptor specific binding | [86,87,89] |
| Ultrasound | Cavitation effect → Mechanical force & Local heating → Carrier depolymerization | UPS | EPR effect & Ultrasound-targeted focusing & Active targeting | Ultrasound intensity, irradiation time, microbubble concentration | Ultrasound precise focusing & Ligand-mediated specific endocytosis | [94] |
| Magnetic | AMF (Alternating Magnetic Field)-induced magnetothermal effect → LCST breakthrough/Carrier depolymerization | UPS/Lysosomal pathway | Magnetic targeting enrichment & Active targeting | Magnetic particle content, magnetic field frequency, temperature rise amplitude | Magnetic field targeting localization & Intracellular high GSH-assisted carrier dissociation | [104] |
| Click reaction | Bioorthogonal click reaction → Stable assembly of nanocomplexes → In situ PROTAC activation | UPS | Metabolic labeling targeting & Ligand-mediated active targeting | Reaction rate constant, metabolic labeling efficiency | Bioorthogonal reaction specificity & Dual targeting | [108,111,112] |
| Enzymatic orthogonal reaction | Orthogonal enzyme-catalyzed substrate hydrolysis → PROTAC precursor activation | UPS | Orthogonal enzyme expression targeting & Ligand active targeting | Enzymatic catalytic efficiency, PROTAC activation rate | Orthogonal enzyme-substrate specific recognition & Ligand-mediated targeted delivery | [113] |
Building on this concept, endogenous self-assembled nanocarrier-mediated TPD systems have substantially expanded microenvironment-responsive strategies. Typically, when exposed to physiological environments, the amphiphilic PROTAC conjugates undergo spontaneous self-assembly driven by hydrophobic interactions and π-π stacking among aromatic moieties, resulting in the formation of stable nanostructures[36]. This self-assembly not only improves the pharmacokinetic stability of PROTACs but also facilitates their cellular uptake via endocytosis. Importantly, intracellular stimuli subsequently destabilize the supramolecular architecture, enabling controlled disassembly and efficient engagement of the E3 ligase - protein of interest (POI) ternary complex[37]. Unlike conventional small-molecule or polymer-based PROTAC systems that depend solely on chemical structure, these self-assembled nanosystems can spontaneously form nanostructures or undergo structural remodeling in response to endogenous signals. This property enables precise drug enrichment, protection, and controlled spatiotemporal release. By exploiting local pH, ROS, GSH, or enzyme levels as triggers, these systems directly link assembly - disassembly processes to pathological signal intensity [Figure 1]. This strategy enhances tissue retention and intracellular delivery while reducing exposure and non-specific toxicity in healthy tissues. Notably, self-assembled structures formed from peptides, proteins, or fusion proteins exhibit high biocompatibility and biodegradability. They form stable nanoaggregates within lesion microenvironments, improving target protein engagement and protecting degradants from plasma enzymatic degradation or lysosomal trafficking during endocytosis[38]. Consequently, endogenous condition-driven self-assembled nano-TPD systems retain microenvironment responsiveness and exhibit distinct advantages in on-demand assembly and signal-amplified release. Endogenous activation, which relies on the TME and physicochemical differences between diseased and normal tissues, enables responses without external stimuli and provides a natural advantage of passive targeting, making it the most extensively investigated activation mechanism.
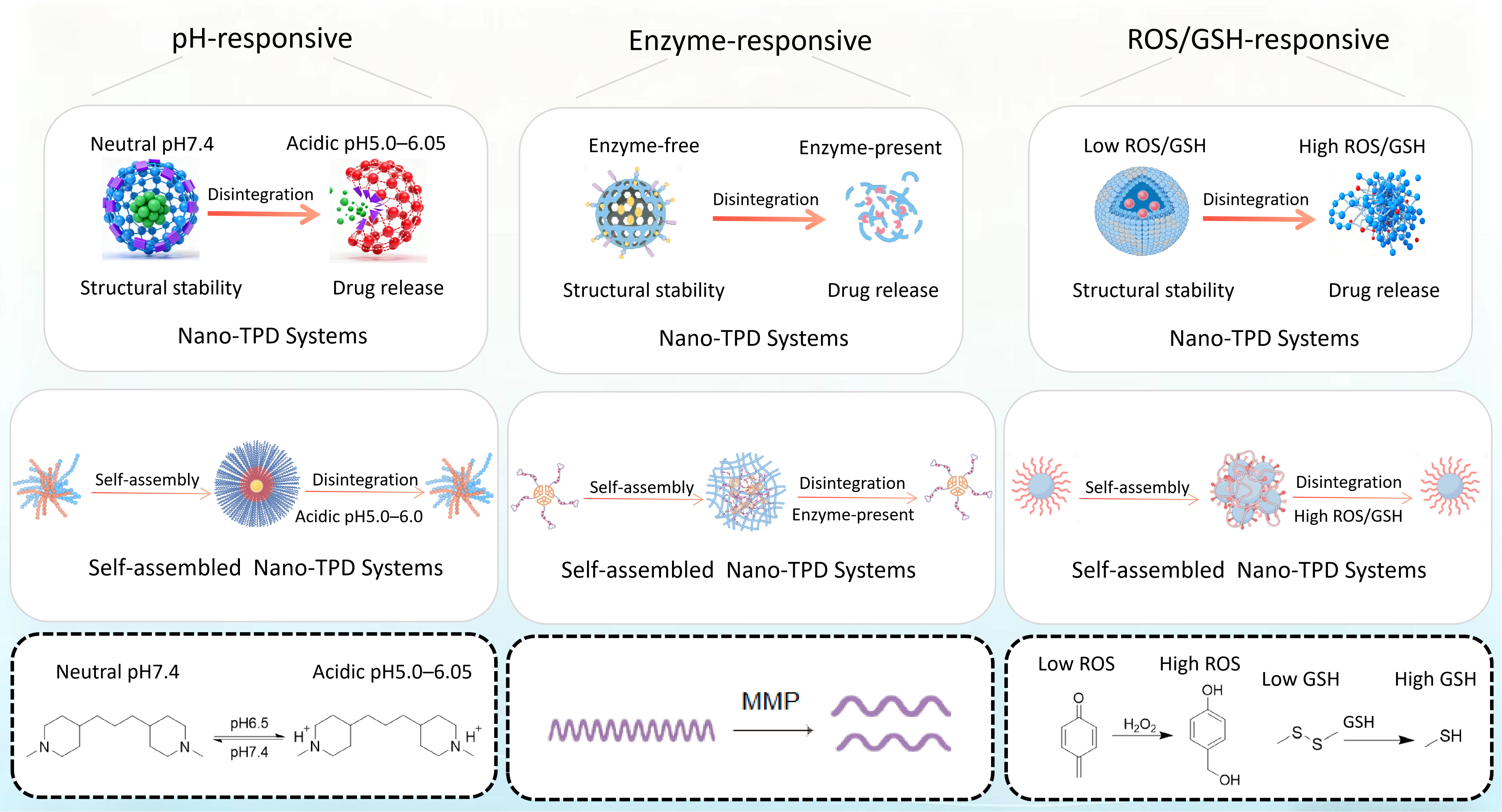
Figure 1. Endogenous condition initiation of nano-TPD systems and self-assembled Endogenous condition initiation of nano-TPD systems. FigDraw ID:UTTRP46ed4. TPD: Targeted protein degradation; ROS: reactive oxygen species; GSH: glutathione; MMPs: matrix metalloproteinases.
pH-responsive
The pH-responsive nano-targeted protein degradation systems typically incorporate target protein degradation molecules into pH-sensitive nanocarriers. These systems remain stable under physiological conditions but undergo structural changes or chemical bond cleavage in acidic environments, enabling controlled release or activation. Building on this foundation, self-assembled pH-responsive nano-targeted protein degradation systems further integrate molecular self-assembly strategies with stimulus-responsive properties, representing a more refined and modular development direction in this field[39]
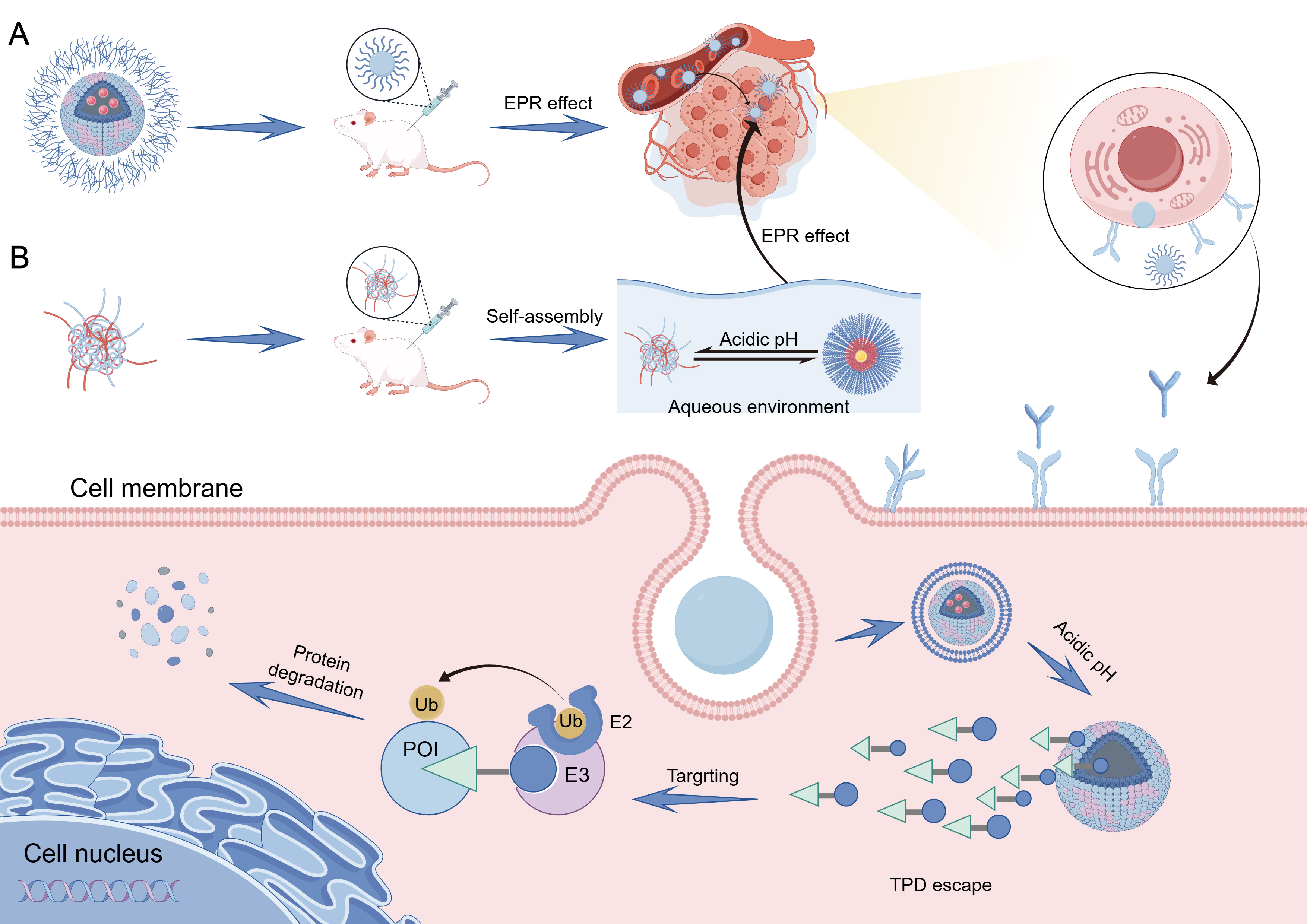
Figure 2. (A) pH-responsive and (B) self-assembled pH-responsive nano-based protein-targeted degradation systems. FigDraw ID:ASTWRe833e. EPR: Enhanced permeability and retention; POI: protein of interest; TPD: targeted protein degradation.
The pH-responsive targeted drug delivery mechanism utilizes the acidic microenvironment of tumors (extracellular pH ~6.5, intracellular pH ~4.5-5.5) as a core activation site. Drug targeting and release occur through three primary mechanisms[42]. First, in polymers with high buffering capacity (e.g., polyamines), proton accumulation within endosomes induces the so-called “proton sponge effect”, in which continuous proton influx is accompanied by chloride ion entry and osmotic swelling. The resulting increase in osmotic pressure leads to endosomal membrane destabilization or rupture, thereby facilitating cytosolic release of degradation molecules[43]. Second, acid-labile bonds (e.g., hydrazide, imine) between drugs and carriers or within carriers hydrolyze under acidic conditions, directly releasing drugs or facilitating intracellular release. Third, PEG detachment strategies responsive to pH utilize PEG coatings to maintain nanocarrier circulation under physiological conditions. In the acidic tumor environment, PEG detaches via acid-sensitive bond cleavage, resolving the “PEG dilemma” and improving intracellular uptake and drug release efficiency at the tumor site[44,45].
The pH-responsive nano-PROTAC developed by the Xue group (MNC@Ca/MnCO3/ARV/anti-PD1) gradually dissolves in the acidic TME (pH ≈ 5.5 - 6.5), releasing internally loaded PROTAC (ARV-825) and anti-PD1 molecules, which specifically degrades BRD4 without damaging immune cells. In neutral tissues (pH ≈ 7.0), the nanocarrier remains stable, minimizing drug release and reducing toxicity to normal cells[46]. The universal nano-ATTEC platform based on mixed-shell polymeric micelles (MSPMs) designed by the Shi team overcomes poor cell-type specificity and dependence on intrinsic autophagy of conventional small-molecule ATTECs. The nano-ATTEC platform self-assembles from three diblock polymers: poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG-b-PCL) modified with ligands for target proteins (e.g., BRD4 or AR), LC3 ligand (ispinesib, IS), and pH-responsive poly(β-amino ester)-b-poly(ε-caprolactone) (PAE-b-PCL). In acidic tumor environments (pH ≈ 6.5), protonation of the PAE segment promotes tumor cell uptake, autophagy activation, and LC3 targeting, leading to protein degradation (e.g., BRD4, AR) via the autophagy-lysosome pathway. Under normal physiological conditions (pH ≈ 7.4), the PEG shell shields the PAE segment, significantly reducing uptake by normal cells[30].
Due to modular design, biocompatibility, and stimulus responsiveness, self-assembled nanocarriers have become ideal platforms for TPD systems (e.g., PROTACs, LYTACs, ATTECs). These nanocarriers self-assemble through non-covalent interactions (e.g., hydrophobic interactions, electrostatic interactions, π-π stacking), encapsulating or covalently conjugating degradation modules[47]. Triggered by specific stimuli, they precisely activate or release degraders, enhancing targeting efficiency and reducing off-target toxicity. Tumors possess lower extracellular pH (~6.5) than normal tissues (~7.4), with even lower lysosomal pH (~4.5-5.0). pH-responsive nano-TPD systems exploit these differences by protonation or cleavage of acid-sensitive groups (e.g., hydrazone, acetal bonds, amino acrylates), releasing degradation modules specifically in tumor tissues[48].
The bifunctional NLTC constructed by the Liu group addresses limitations such as insufficient tumor accumulation, non-specific activation, and low monotherapy efficacy. The lysosome-targeting bifunctional chimeric nanoplatform self-assembles into a nanocapsules structure with surface-modified lysosome-targeting ligands. Upon entering cells, protonation of amino groups in acidic lysosomes triggers structural dissociation, releasing LYTAC degraders. These degraders specifically degrade tumor cell surface proteins and activate antitumor immune responses. This system demonstrated a tumor inhibition rate of 87.9% in a 4T1 breast cancer model[31]. To address the limitations of traditional LYTACs in hepatocellular carcinoma (HCC) therapy, such as low bioavailability and nonspecific distribution, the Chao group developed an intelligent modular DNA LYTAC (IMTAC) nanodevice and a drug EGFR/PDL1 IMTACs. Using self-linked DNA nanocircles as the carrier, this system achieves specific activation in the TME (pH 6.5-7.0) by precisely regulating the stoichiometry and modular distribution of pH-responsive degradation promoters (targeting insulin-like growth factor II receptor, IGFIIR) and EGFR/PDL1-targeting ligands, with an optimal spacing of 20 nm. The nanodevice enables synergistic degradation of EGFR and PDL1, reducing EGFR levels to approximately 10% in HepG2 cells in vitro and achieving a tumor inhibition rate of 80% in vivo, without significant toxic side effects[49]. The Qi group has developed a novel ionizable nano-PROTAC (i16-ETNc) platform, which co-assembles a pH-responsive tertiary amine and amphiphilic conjugates. This combination enhances cellular uptake and endosomal escape via the proton sponge effect, overcoming traditional nano-PROTAC endosomal entrapment. The platform achieves potent degradation of leucine-rich α-2-glycoprotein 1 (LRG1) in cells (DC50 = 17.1 μM and 19.5 μM for 4T1 and MDA-MB-231 cells, respectively). In vivo, it accumulates in tumors through EPR effect and active targeting, prolongs blood circulation (t1/2 = 1.27 h), reduces tumor weight by 71.3%, and demonstrates no significant systemic toxicity, offering a universal strategy for TPD[50].
This system’s advantages include its targeting capability and biocompatibility, it activates only under acidic conditions in diseased tissues or organelles, minimizing harm to normal tissues. Moreover, most acid-sensitive bonds comprise biodegradable materials, ensuring low toxicity of degradation products. However, substantial challenges remain, such as significant intratumoral pH gradient variations requiring precise control of acid-sensitive bond cleavage thresholds; endosomal escape efficiency affecting intracellular delivery and degradation by lysosomal enzymes; and inconsistent responses caused by pH fluctuations across patients or lesions.
Enzyme-responsive
The tumor microenvironment is frequently characterized by an aberrant protease landscape, in which multiple matrix metalloproteinases (MMPs) are upregulated in a wide range of tumor tissues and their surrounding extracellular matrix, and are closely associated with tumor invasion, metastasis, and disease progression[51,52]. Enzyme-responsive nano-targeted protein degradation systems are typically designed by exploiting the abnormal expression and/or activity of tumor-associated enzymes - such as MMPs and cathepsins - within tumor tissues or along cellular endocytic pathways. In these systems, enzyme-cleavable substrates or linkers are incorporated into degrader prodrugs or nanocarrier architectures; upon encountering the target enzymes, site-specific cleavage occurs, thereby triggering the release and/or activation of degraders[53].
The Ryu team developed a cancer-specific carbonic anhydrase IX (CAIX)-targeting supramolecular nanofibrous lysosome-targeting chimera (Supra-LYTAC). It comprises two hetero-functionalized, self-assembling amphiphilic Phe-Phe-Lys (FFK)-based peptides: one targets CAIX expressed on cancer cell membranes, and the other interacts with the POI. The supramolecular nanofibers near CAIX form a ternary complex (CAIX-nanofiber-POI). These complexes undergo clathrin-mediated endocytosis into lysosomes, where the acidic hydrolytic enzyme-rich environment facilitates the degradation of POIs[54].
Tumor tissues overexpress specific proteases [e.g., MMPs, cathepsins (Cts), and plasmin][55]. Enzyme-responsive self-assembled nano-TPD uses protease-sensitive peptides as linkers or assembly units. Upon specific enzymatic cleavage at tumor sites, carriers undergo depolymerization or degradation module activation, enabling precise control.
The Gao team constructed dBET6@CFMPD nanomedicine by self-assembling MMP-2-sensitive peptide (FFRFKGPLGLAGC)-PEG-DSPE conjugates modified with the photosensitizer Ce6 (chlorin e6) and the BRD4-targeting PROTAC (dBET6). This system addresses multiple challenges: poor PROTAC solubility, insufficient tumor accumulation, limited single-agent therapy efficacy, immune escape caused by PD-L1 (programmed death-ligand 1) upregulation after photodynamic therapy (PDT), and impaired nanoparticle penetration and retention. At tumor sites, the highly expressed MMP-2 triggers structural transformation from spherical to nanofibrous morphology, enhancing Ce6 retention, PDT efficacy, and immunogenic cell death (ICD). Simultaneously, released dBET6 degrades BRD4, downregulates c-Myc and PD-L1 expression, reprograms tumor-associated macrophages (TAMs), and synergistically inhibits primary breast tumors and lung and brain metastases[56].
The split-and-mix liposomal PROTAC system (LipoSM- PROTAC) self-assembles from folate (FA)-modified liposomes. Through DSPE-PEG2000, it links the E3 ubiquitin ligase ligand (VHL) and ERα-targeting ligand (4-hydroxytamoxifen, Tam), with MMP-2-sensitive peptides on the surface. Following MMP-2 cleavage at tumor sites, the liposome induces synergistic ligand activity, selectively degrading ERα protein and effectively inhibiting MCF-7 breast cancer cell growth[57]. The targeted biomolecule regulation platform (SM-PROTAC) self-assembles into nanospheres with functional modules via diphenylglycine peptides (δδRR), encapsulating split PROTAC fragments (E3 ligand module and POI ligand module). After cellular uptake, the modules form active PROTACs in situ, targeting multiple proteins [e.g., ERα, cyclin-dependent kinases 4/6 (CDK4/6), bromodomain-containing proteins 2/4 (BRD2/4)] without significant activation in normal tissues[58].
Enzyme-catalyzed processes exhibit amplification effects, as one enzyme molecule can cleave multiple carrier molecules, significantly enhancing sensitivity. Additionally, carriers enable complete control from stable circulation and tumor accumulation to stimulus-triggered release and degrader activation, reducing non-specific in vivo release and associated toxicity. However, enzyme activity may vary significantly among individuals, disease states, and tumor/inflammatory lesions. Thus, optimizing enzyme cleavage site sensitivity, carrier stability, and release kinetics based on actual enzyme expression remains critical. Furthermore, synthetic complexity, large-scale production, and quality control present barriers to clinical translation of enzyme-responsive nano-TPD. Although preliminary in vivo data demonstrate efficacy, systemic safety, localization and degrader release efficiency in complex microenvironments, and long-term effects still require comprehensive evaluation.
ROS/GSH-responsive
Under normal physiological conditions, intracellular redox reactions are typically maintained in a state of equilibrium[59]. However, tumor tissues experience disturbances in oxidizing and reducing agents due to environmental factors and signal regulation, leading to redox imbalance. These disturbances subsequently influence tumor cell behavior, including metabolism, proliferation, metastasis, and drug resistance[60].
ROS represent the main oxidizing agents inside cells. ROS are mainly generated by the mitochondrial respiratory chain during oxidative stress. Common ROS include hydroxyl radicals (•OH), superoxide anions (•O2-), singlet oxygen (1O2), and H2O2[61]. Under normal conditions, ROS remain at low levels (0.02-1 μM), supporting normal cell growth and apoptosis. However, when ROS levels significantly rise (50-100 μM) ,proteins associated with ROS-sensitive signaling pathways become activated, leading to oxidative stress in tumors or inflamed tissues[62]. The main reducing agents within cells include GSH, Catalase (CAT), thioredoxin, and Fe2+. Among these, GSH has the highest cytoplasmic concentration in tumor cells (2-10 mM), greatly exceeding its extracellular concentration (2-20 μM)[63]. GSH concentration in tumor cells is about 1000-fold higher than in plasma and at least four times greater than in normal tissues[64].
Chen’s group developed ROS-responsive Pre-PROTACs, a type of ROS-responsive small-molecule conjugate, by introducing ROS-cleavable groups (aryl boronic acid) into the parent PROTAC molecules. These Pre-PROTACs efficiently degrade the target protein BRD3 in response to elevated ROS levels[65]. Bi et al. designed a ROS-responsive nano-PROTAC (BT-NanoPROTAC) targeting BRD4, which is formed by the self-assembly of Small-Molecule Conjugate BT-PROTAC (ARV771 modified with a boronic acid group) and Poloxamer 188 into nanoparticles and remains stable in the bloodstream. This system leverages the high ROS levels in activated hepatic stellate cells (aHSCs) to release ARV771, a potent BRD4 degrader. The BT-NanoPROTAC specifically degrades BRD4 in aHSCs, downregulating fibrosis markers such as α-SMA and type I collagen, thus demonstrating therapeutic potential for hepatic fibrosis both in vitro and in vivo[66]. The ROS-guided supramolecular assembly system (TCO-MZ1) contains a PROTAC precursor and ROS-sensitive monomers. In the ROS-rich tumor environment, oxidative reactions occur in these monomers, reorganizing micelles into more stable nanofiber structures and prolonging PROTAC retention in tumors. Concurrently, ROS triggers precursor activation, degrading PD-L1 and reversing the immunosuppressive microenvironment[67].
The redox balance in tumor cells is disrupted, with GSH concentrations (10-20 mM) significantly higher than normal tissues (2-10 μM), accompanied by substantially elevated ROS levels. ROS/GSH-responsive self-assembled nano-TPDs employ sensitive linkers, such as disulfide and selenoether bonds, which break upon changes in the redox environment to release PROTACs. Glutathione-scavenging nanoparticles (ARV@PDSA Nano-PROTAC) possess a core-shell structure created by polymer self-assembly. The core encapsulates PROTAC agents, and the shell features a GSH-sensitive cross-linked disulfide network. Upon entering tumor cells, high intracellular GSH concentrations cleave the disulfide bonds, triggering carrier disassembly and PROTAC release. Simultaneously, these nanoparticles consume excess GSH, increasing oxidative stress and synergistically enhancing HER2 degradation. This approach achieves a 40% higher tumor growth inhibition rate compared to PROTAC treatment alone[68]. To address the broad-spectrum cytotoxicity issues associated with molecular glues, the Yuan team designed a targeted molecular glue, H1-mGlu, against the Bcr-Abl protein, and constructed a self-assembling nanocarrier, Cle-NP. This system triggers drug release through high concentrations of GSH/H2O2 in the tumor microenvironment, achieving tumor-specific degradation. In vitro experiments demonstrated that Cle-NP exhibited a median inhibitory concentration (GI50 = 74.8 nM) for K562 cells, effectively degraded Bcr-Abl (DC50 = 0.89 μM), and induced apoptosis. In vivo, Cle-NP significantly inhibited tumor growth (TGI = 78.1%) in K562 xenograft models without causing notable liver damage or weight loss, resolving the systemic toxicity issues of traditional molecular glues and providing a novel strategy for precision cancer therapy[29]. The self-assembled LYTAC nanosystem connective tissue growth factor (CTGF)-LYTAC (NanoCLY) is a GSH-responsive nanoparticle self-assembled from amphiphilic chimeras, which targets CTGF by forming nanoaggregates through peptide chain hydrophobic interactions, with tumor-targeting peptides on its surface. Upon entering the GSH-rich intracellular environment, the disulfide bonds linking the hydrophilic CL8-M6P and the hydrophobic lauryl group are cleaved, disrupting the amphiphilic balance of the chimeras and leading to the disassembly of the originally self-assembled spherical nanostructures, thereby releasing the functional drug molecule CL8-M6P. This process mediates CTGF degradation and remodels the inflammatory TME, significantly enhancing the therapeutic efficacy against triple-negative breast cancer[69].
ROS-responsive nano-TPD systems are characterized by rapid response and high selectivity. However, ROS molecules have short half-lives in vivo (e.g., approximately milliseconds to seconds for H2O2). Additionally, variability in ROS types among different lesions (e.g., H2O2 predominant in tumors, superoxide anions predominant in inflammation) may impact carrier response efficiency and TPD[70,71].
Overall, endogenous condition - initiated nano-TPD systems exploit intrinsic physicochemical and biochemical disparities between diseased and normal tissues to achieve passive targeting and stimulus-responsive activation without external inputs. Owing to their design simplicity and biological relevance, this activation mode remains the most extensively investigated strategy in nano-TPD research. However, its reliance on heterogeneous endogenous signals also highlights inherent limitations in controllability and activation precision, motivating the exploration of exogenously triggered and multi-condition - initiated nano-TPD systems discussed in the following sections.
EXOGENOUSLY INITIATED NANO-TPD SYSTEMS
Endogenously triggered TPD (especially prodrug/nanodelivery-based degradation systems activated by tumor-endogenous signals) is susceptible to the complexity and heterogeneity of the tumor microenvironment, interpatient variability, and tumor evolution, leading to uncertainties in activation efficiency and site-specificity. Meanwhile, PROTAC molecules themselves generally face challenges such as insufficient cell membrane permeability and limited in vivo exposure/stability, rendering precise regulation after administration relatively difficult. In contrast, exogenous triggering relies on externally applied non-physiological signals (e.g., light, ultrasound, magnetic fields, bioorthogonal reactions) to induce responses, offering advantages of remote controllability and high spatiotemporal precision. This approach can partially enhance the selectivity of the degradation process and reduce the risk of systemic toxicity[72] [Table 2].
The integration of exogenous stimulus-responsive materials with PROTAC technology can further minimize off-target exposure through nanodelivery and “activatable/releasable” designs, improve stability in biological media, enable on-demand therapy, and enhance cellular uptake and transmembrane delivery efficiency[73]. Based on the physicochemical properties of the triggering signals, exogenous stimuli are generally categorized into two major types: physical signal-triggered and chemical signal-triggered[74] [Figure 3].
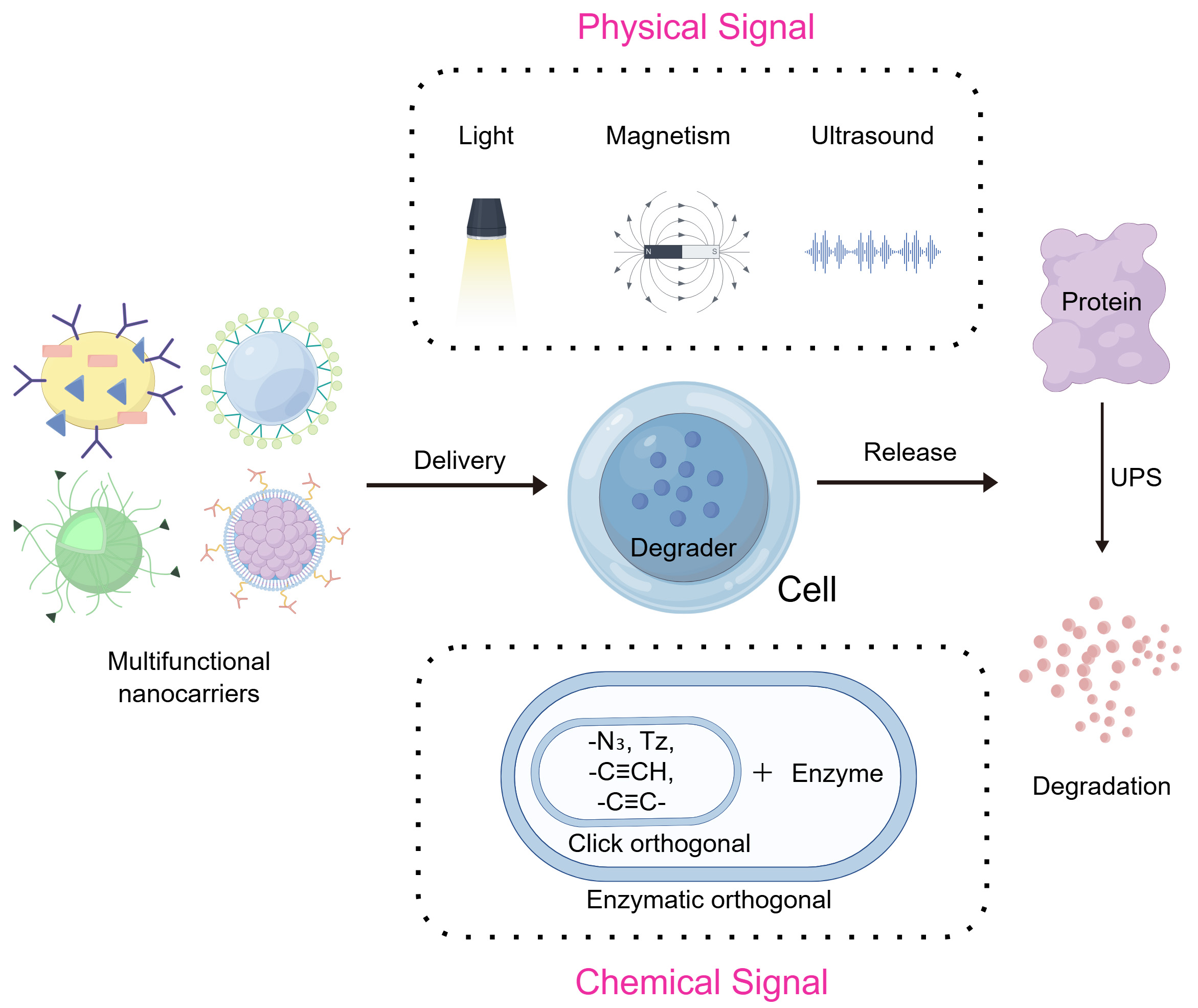
Figure 3. Exogenous signals triggering the nano-TPD systems. FigDraw ID:ISAOU2d242. TPD: Targeted protein degradation; UPS: ubiquitin-proteasome system.
Exogenously initiated self-assembled nano-TPD systems have recently attracted growing attention. In these platforms, PROTAC conjugates or functional building blocks form supramolecular nanostructures through noncovalent interactions, ensuring structural stability during systemic delivery. Upon exposure to an external stimulus, such as light, Magnetic or ultrasound, the supramolecular assembly undergoes structural rearrangement or disassembly, leading to rapid PROTAC activation or release[75,76]. This stimulus-driven assembly - disassembly behavior enables precise temporal control over protein degradation and allows degradation to be initiated only at the desired anatomical site.
Physical signal activation
Physical signal activation represents a pivotal strategy that triggers system responses through externally applied physical energy (e.g., light, ultrasound, or magnetic fields). Compared with endogenous microenvironmental response mechanisms, these systems are immune to individual variations or pathological heterogeneity, demonstrating excellent predictability and reproducibility[77]. Furthermore, physical signal stimulation offers distinct advantages including non-invasiveness, remote control capability, and precise parameter adjustment. This enables researchers to initiate or enhance protein degradation processes at specific temporal and spatial locations, achieving high-precision modulation of target protein degradation. Such highly controllable stimulation methods provide novel opportunities to minimize systemic side effects and optimize therapeutic windows.
Light-responsive Systems
The non-invasive and tunable properties of light (intensity and wavelength) facilitate remote spatial-temporal control of photosensitive nanomaterials at targeted sites in vivo. This has led to their widespread adoption in biomedical applications[78,79] [Figure 4].
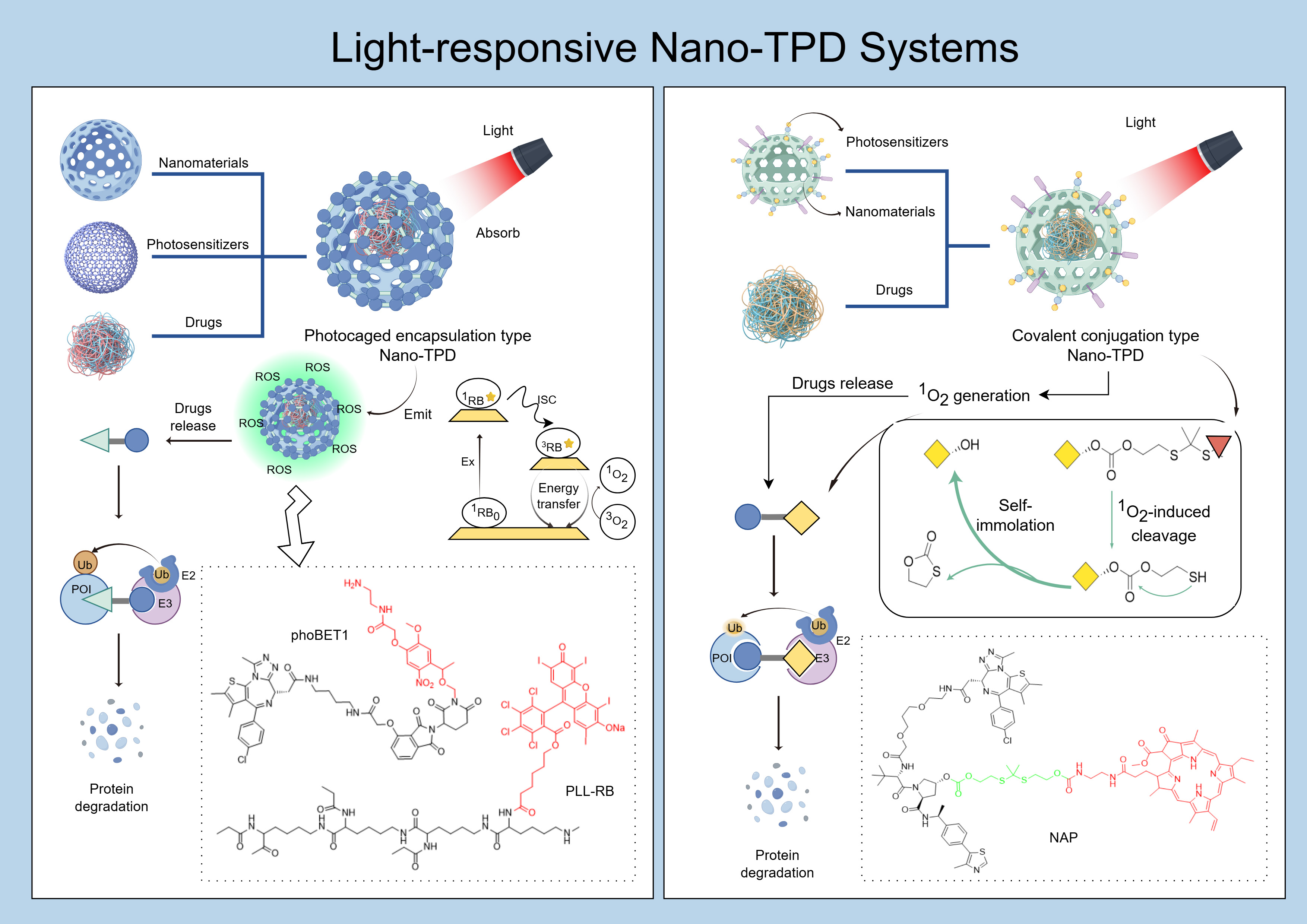
Figure 4. Light-responsive nano-TPD systems. FigDraw ID:WIIPU29991. TPD: Targeted protein degradation; POI: protein of interest; ROS: reactive oxygen species.
Primary light-mediated therapies include photothermal therapy (PTT) and PDT [80]. PTT employs photothermal conversion agents (PTCAs) to convert light into heat, achieving localized cancer ablation[81]. PDT activates photosensitizers using safe visible light. These photosensitizers interact with substrates, such as cell membranes, generating free radicals or directly reacting with oxygen to produce cytotoxic ROS. This induces cellular stress and death responses in tumors[82,83]. Most photosensitizers are responsive to ultraviolet (UV) and visible light; however, limited penetration depth and potential phototoxicity restrict their clinical applications. Thus, near-infrared (NIR) light, with deeper penetration and reduced toxicity, has gained attention[84,85].
Combining photosensitizers with PROTAC technology generally employs two approaches. The first method involves encapsulating PROTAC molecules within photocages containing photosensitizers. Upon targeted illumination, these photocages degrade, releasing PROTACs at specific sites. Additionally, phototherapy enhances the degradation efficiency against targeted tissues[86-88]. The Zhang research group develop a near-infrared light-controlled PROTAC delivery device (USDPR) by encapsulated BRD4-targeting PROTACs (dBET6) within hollow silica-coated upconversion nanoparticles (UCNPs). They then coated these nanoparticles with a cationic polymer composed of polylysine covalently linked to the rose bengal photosensitizer. This cationic polymer transfers energy from UCNPs to the photosensitizer, generating ROS, disrupting cell membranes, facilitating PROTAC entry into the cytoplasm, and preventing entrapment in endosomes or lysosomes[86]. The Yu research group loaded photocaged PROTAC (phoBET1) into UCNP-based mesoporous silica nanoparticles (UMSNs), forming a NIR light-activatable PROTAC nanocage (UMSNs@phoBET1). Under NIR irradiation, UCNPs convert NIR into UV light, removing the photocage from PROTAC and releasing its active form,thereby degrading bromodomain-containing protein 4 (BRD4) and inducing apoptosis in MV-4-11 cancer cells[87].
The second approach directly conjugates a photosensitizer to the PROTAC through a cleavable linker. Upon light exposure, the ROS generated by the photosensitizer cleaves the linker, releasing active PROTAC. The released ROS also induces apoptosis in targeted cells, enhancing antitumor effects[89,90]. The Li research group conjugated the PROTAC (ARV-771) with a NIR photosensitizer (pheophorbide a, PA) using a self-immolative thioketal linker[89]. Under NIR irradiation, PA generates 1O2, cleaving the linker to release PROTAC while simultaneously performing PDT. Light-responsive activation leverages the penetration depth and precise control of specific wavelengths (particularly NIR light) to activate photosensitizers, generating ROS or triggering photoisomerization. This leads to nano-TPD depolymerization or PROTAC activation, enabling accurate spatial-temporal regulation of protein degradation. The self-assembled nano-PROTAC system is formed by covalently linking the NIR photosensitizer (pheophorbide a) with a BRD4-targeting PROTAC (ARV-771) through a self-immolative thioketal linker, subsequently self-assembling into nanoparticles (NAP). Following intravenous injection, NAP accumulates at tumor sites due to EPR effect. Under 670 nm NIR irradiation, PA generates 1O2, which cleaves the linker, releasing PROTAC and inducing apoptosis of tumor cells. The released PROTAC degrades BRD4 protein, achieving an 83.4% tumor inhibition rate in combination with PDT.
Ultrasound-responsive systems
Although near-infrared light-activated targeted degradation drugs have been developed for safer and more effective tumor therapy, limitations remain, including shallow tissue penetration (less than 1 cm) and potential phototoxicity[91,92].Conversely, ultrasound possesses greater tissue penetration (more than 10 cm) and provides safe, non-ionizing radiation. Its penetration depth can be precisely regulated by adjusting frequency, duty cycle, and exposure duration[76,91,93,94]. Thus, ultrasound-mediated targeted drugs have attracted significant attention. Studies suggest that ultrasound-mediated sonodynamic therapy (SDT) operates through two primary mechanisms. The first mechanism is cavitation, whereby ultrasound irradiation causes cavitation nuclei in liquids to undergo expansion, contraction, and rupture, producing high temperatures and pressures. This process leads to pyrolysis and generates ROS. The second mechanism involves ultrasound luminescence. Here, cavitation bubbles emit light upon ultrasound irradiation, activating sonosensitizers to form electron-hole pairs and generate ROS[92,93,95,96].
Researchers have combined SDT with LYTAC-mediated immunotherapy. The Pu research group designed a targeted polymeric nano-lysosome-targeting chimera (SPNly)[94]. This construct comprises a semiconducting polymer backbone (PCPDTODBT) conjugated via polyethylene glycol (PEG) linkers with interleukin-4 receptor (IL-4R)-targeting peptide (IL4Rp) and lysosome-sorting peptide (LSP) moieties. Ultrasound irradiation induces sonoluminescence in the 350-650 nm wavelength range, activating nano-LYTAC to generate 1O2, thereby causing ICD of tumor cells. Nano-LYTAC also targets IL-4R, highly expressed on M2 macrophages, and mediates IL-4R degradation via the endosome-lysosome pathway. Consequently, nano-LYTAC modulates M2 macrophage function in a dose-dependent manner, reducing protumor markers (CD206 expression and IL-10 secretion) at lower concentrations and inducing apoptosis at higher concentrations. Additionally, nano-LYTAC exhibits prolonged retention in tumors (> 48 h), enabling multiple rounds of SDT from a single administration.
Magnetic-responsive systems
Due to inherent magnetism, superparamagnetism, and low toxicity, magnetic nanocarriers have broad biomedical applications[97]. Under a static magnetic field, magnetic nanoparticles can achieve magnetic targeting - mediated accumulation, thereby modulating the in vivo distribution and localized deposition of therapeutic agents. In contrast, exposure to an alternating magnetic field enables magnetic hyperthermia, which induces localized temperature elevation and, in certain systems, enhances oxidative stress or reactive oxygen species generation, ultimately leading to therapeutic sensitization or stimuli-triggered drug release[98-100]. In addition, magnetic nanomaterials can serve as magnetic resonance imaging (MRI) contrast agents or responsive magnetic resonance (MR) nanoprobes for tumor imaging and theranostic applications[101]. Furthermore, magnetic nanoparticles can synergistically combine EPR-based passive accumulation with externally applied magnetic field - guided targeting to further enhance drug accumulation in tumor tissues, and this effect can be augmented by ligand-mediated active targeting strategies to improve targeting efficiency[102,103].
Currently, research on magnetic nanocarriers conjugated with PROTAC remains limited. Sun’s group sequentially administered two therapeutic agents within the TME. First, biodegradable calcium-doped manganese carbonate-coated magnetic nanoclusters (MCM) loaded with BRD4-targeted PROTAC (ARV-825, forming MCM/ARV) were delivered. This was followed by administration of magnetic nanocluster (MNC)-modified bone marrow-derived dendritic cells (M-BMDCs). This strategy increased dendritic cell (DC) presence in the TME and magnetized tumor-associated antigens (TAAs) released by PROTAC-induced tumor ICD. Magnetized TAAs then attracted M-BMDCs, significantly improving antigen uptake and DC activation, thereby initiating potent antitumor immune responses[104].
Chemical signal activation
Chemically activated nanoscale protein degradation systems primarily utilize exogenous bioorthogonal reactions as their trigger mechanism. By introducing bioorthogonal chemical reagents or specific enzyme-substrate pairs, these systems induce highly targeted chemical reactions within complex biological systems to activate the degradation process. As bioorthogonal reactions do not interfere with endogenous biochemical processes, this strategy demonstrates distinct advantages including high reaction specificity and excellent biocompatibility, enabling precise and controlled protein degradation in cellular or in vivo environments[105].
Currently, chemical signal activation strategies primarily fall into two categories: click chemistry-based approaches and enzyme-catalyzed orthogonal reactions. The former utilizes classical or modified click chemistry reactions to achieve in-situ assembly or conformational transformation of target molecules, while the latter relies on substrate conversion mediated by specific enzymes to enable spatiotemporal control of degradation activity[106]. These strategies provide crucial tools for constructing highly selective nanoscale protein degradation systems in complex biological environments.
Click reaction
Click chemistry triggers cycloaddition reactions by externally introducing orthogonal reagents [e.g., azide, Dibenzocyclooctyne (DBCO), tetrazine][107]. This method enables in situ activation or assembly of nanocarriers and provides precise control over the release of degradation modules. Huang et al. developed a microneedle-assisted, click chemistry-activated PROTAC system. Tetrazine (Tz) was encapsulated within dissolvable microneedles, and a PROTAC prodrug (MZ1-O) was loaded into poly (lactic-co-glycolic acid) (PLGA) nanoparticles (MZ1-O@NP)[108]. Following microneedle-mediated local Tz release at the tumor site, Tz reacts with intravenously administered MZ1-O@NP via inverse electron-demand Diels-Alder (IEDDA) click chemistry. This reaction removes the prodrug’s protective group, activating MZ1. The active MZ1 subsequently degrades BRD4 protein, significantly inhibiting tumor growth.
Notably, bioorthogonal click reactions such as Strain-Promoted Azide-Alkyne Cycloaddition (SPAAC) and IEDDA offer highly selective and biocompatible activation mechanisms, allowing nano-PROTAC systems to achieve spatiotemporally controlled protein degradation through externally triggered in situ prodrug conversion or supramolecular assembly[109,110]. The bioorthogonal chemistry-based, in vivo self-assembling PROTAC system (Nano-CLIPTACs) comprises nano-precursors prepared by separately encapsulating trans-cyclooctene-tagged (TCO-tagged) protein-of-interest (POI) ligands (POI-TCO) and tetrazine-tagged pomalidomide derivatives (POM-TZ) into cRGD-modified liposomes. After intravenous co-administration, the nano-precursors accumulate at the tumor site via EPR effects, triggering in situ PROTAC formation through IEDDA click chemistry. These assembled PROTAC molecules specifically degrade anaplastic lymphoma kinase (ALK). In an ALK-positive H3122 lung cancer xenograft model, the high-dose group achieved a 77.4% tumor inhibition rate[111]. Bioorthogonal POLY-PROTAC nanoparticles were prepared via PEG-induced RAFT copolymerization, resulting in amphiphilic polymers with azide-terminated groups. PROTAC precursors (Me-ARV771/Me-MZ1) were grafted onto polymer backbones via disulfide bonds, forming micellar nanocarriers through self-assembly. Following intravenous injection of DBCO-modified pretargeting nanoparticles (PED NPs), the NPs dissociate within the acidic TME, exposing DBCO groups. Subsequently administered azide-modified POLY-PROTACs react with exposed DBCO groups, cross-linking and solidifying the carriers, thus extending their retention at tumor sites. Simultaneously, PROTAC precursors are released by GSH-mediated disulfide cleavage in tumor cells, specifically degrading bromodomain-containing protein 4 (BRD4). This approach exhibits favorable biocompatibility, with no significant weight loss or organ pathology observed during treatment[112].
Enzymatic orthogonal reactions
Enzymatic orthogonal reactions regulate TPD by artificially introducing orthogonal enzymes or substrates minimally expressed in normal tissues, specifically activating degradation modules at tumor sites.
The Xing research group developed enzyme-activated clickable PROTAC (ENCTACs). These consist of a pomalidomide derivative (Module 1, Y952) bearing a cathepsin B (CTSB)-cleavable Val-Cit peptide bond and a GSH-responsive -StBu protective group, and a JQ1 derivative (Module 2, J558) modified with 2-cyanobenzothiazole (CBT). Both small-molecule fragments are administered intravenously, reaching the tumor site. Highly expressed CTSB at the tumor site cleaves the Val-Cit peptide, exposing an amino group. Additionally, high intracellular GSH concentrations reduce and remove the -StBu protective group, releasing a free thiol group. This thiol subsequently reacts with CBT from Module 2 through an enzymatic orthogonal condensation reaction. The assembled PROTAC specifically degrades BRD4, downregulates PD-L1 expression, and significantly enhances infiltration of CD4+ and CD8+ effector T-cells into tumor tissue[113].
Collectively, while exogenously initiated nano-TPD systems markedly improve controllability and precision compared with endogenous strategies, their dependence on single external triggers highlights the need for more sophisticated activation paradigms. These considerations have driven the emergence of multi-condition - responsive nano-TPD platforms, in which endogenous pathological cues are integrated with exogenous control signals to achieve logic-gated, highly selective, and safer protein degradation, as discussed in the following section.
MULTI-CONDITIONALLY RESPONSIVE NANO-TPD SYSTEMS
Despite significant advances in both endogenous- and exogenously initiated nano-TPD platforms, the clinical translation of PROTAC-based therapies remains hampered by insufficient tumor specificity, residual off-target degradation, and pronounced heterogeneity of the TME. Single-trigger activation - whether relying solely on endogenous pathological cues or externally applied physical stimuli - often fails to simultaneously achieve high biological specificity and precise spatiotemporal controllability[114-116]. These limitations have driven the development of multi-conditionally responsive nano-TPD systems. Multi-condition-responsive nano-targeted degradation systems combine two or more activation signals (e.g., endogenous-endogenous, endogenous-exogenous, or exogenous-exogenous) to construct sequential response or multiple stimulus response mechanisms. This enables controlled release and/or activation of degradants upon specific signal combinations, thereby enhancing spatiotemporal selectivity in protein degradation while potentially reducing systemic toxicity[117].
Currently, nano-PROTAC spatiotemporal control strategies predominantly involve “single exogenous + single endogenous” or “dual endogenous” activation. A single exogenous signal (e.g., light, ultrasound, or magnetic fields) already provides centimeter-level spatial resolution at tumor sites. Coupling with endogenous TME-derived signals (e.g., pH, enzyme, or ROS/GSH) sufficiently achieves precise activation[118]. Although integrating a second exogenous physical stimulus may further reduce off-target effects, precise in vivo alignment of two physical fields substantially increases risks related to therapeutic windows, positioning accuracy, and consistency. Furthermore, PROTACs irreversibly degrade target proteins via the UPS, while nanocarriers typically mediate PROTAC release through lysosomal pathways. Considering the intrinsic high activity of PROTAC molecules, researchers prioritize preventing false activation over introducing additional gating components[119]. Additionally, dual-exogenous responsiveness necessitates integrating nanocarriers, PROTACs, dual-responsive moieties, and potential imaging agents. Such complexity presents significant barriers to scale-up production, batch reproducibility, metabolic analysis, and safety evaluation[120]. To date, no in vivo proof-of-concept study for “dual-exogenous activation nano-PROTACs” has been reported, with related discussions still at a theoretical stage. Thus, based on recent progress, this review elaborates on combined activation systems from two perspectives: “dual endogenous activation” and “endogenous-exogenous activation” [Figure 5].
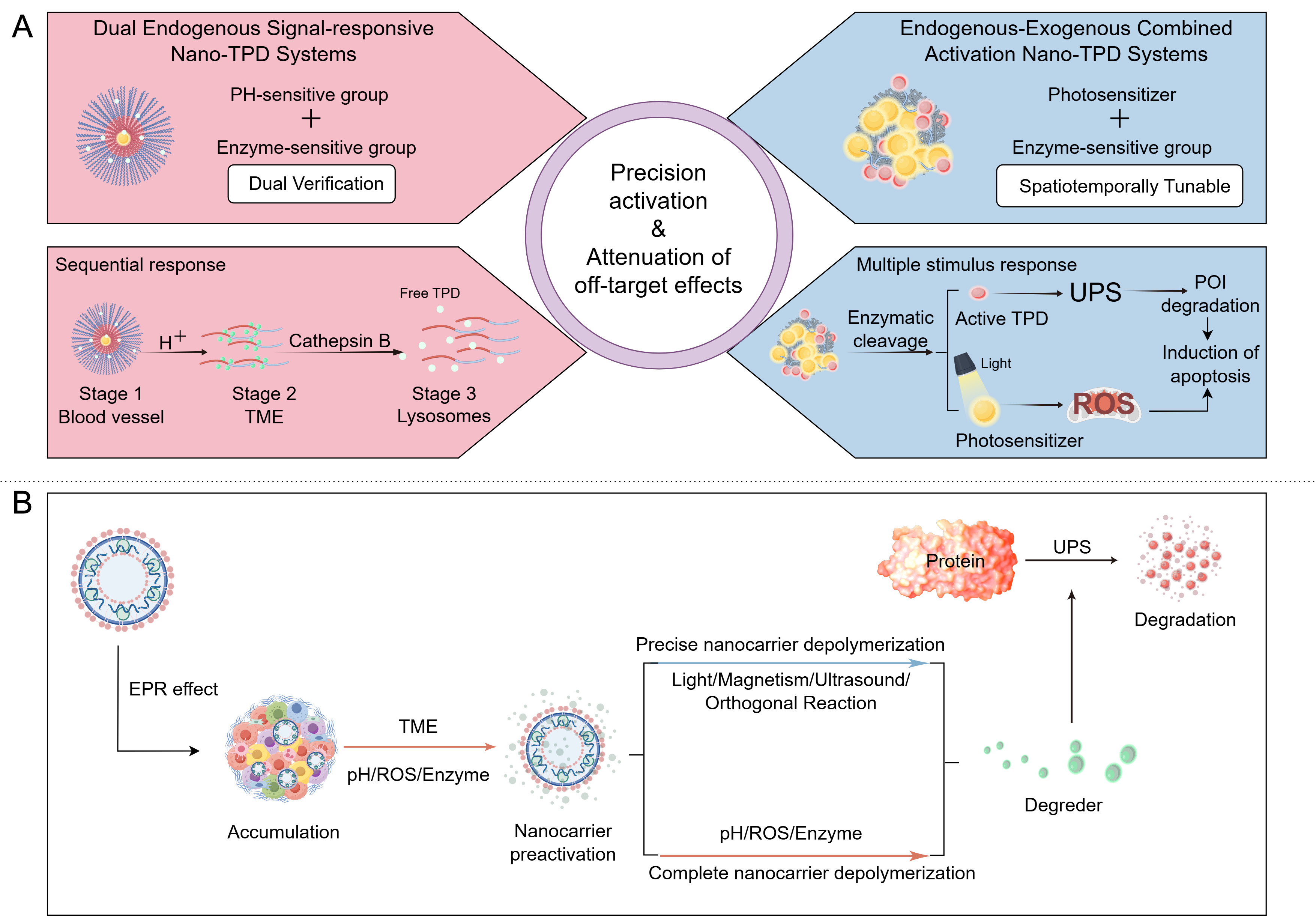
Figure 5. Schematic of precise activation strategies and delivery-degradation process for nano-TPD systems. (A) Left panel: Responsive conditions and responsive process of the Dual Endogenous Signal-responsive nano-TPD Systems; Right panel: Responsive conditions and responsive process of the Endogenous-Exogenous Combined Activation nano-TPD Systems; (B) Overall flowchart of the Multi-conditionally responsive nano-TPD Systems. FigDraw ID:TUTPRf5a0d. TPD: Targeted protein degradation; UPS: ubiquitin-proteasome system; POI: protein of interest; ROS: reactive oxygen species; EPR: enhanced permeability and retention; TME: tumor microenvironment.
Dual endogenous signal-responsive systems
Dual-endogenous co-activation systems target two distinct pathological signals within the TME, including pH, enzyme activity, ROS, GSH, and copper ions[121,122]. Leveraging synergistic effects of these signals provides dual verification, ensuring degradation initiation only occurs at the intended site. This strategy is particularly effective for malignancies characterized by clearly defined microenvironmental profiles, such as pancreatic and lung cancers[123].
Combination of pH and enzyme or GSH signals
Sequential response to pH and enzyme signals represents a classical design within this category. Yang et al. developed pH/CTSB sequential-responsive nanoparticles (PSRNs) based on the acidic TME (pH 6.5-7.0) and high CTSB expression in colorectal cancer (CRC) lysosomes. The system employs block copolymer PEG-b-P(EPA-r-PROTAC) as the carrier, conjugating CDK4/6-targeting PROTAC to the copolymer backbone via the CTSB-sensitive tetrapeptide (GFLG). Under physiological conditions (pH 7.4), PSRNs maintain a stable nanoparticle structure (~40 nm) to prolong blood circulation. Upon entering acidic tumor regions, EPA chains become protonated, causing nanoparticle dissociation into smaller unimers (< 10 nm), facilitating tumor penetration. Ultimately, CTSB cleaves the GFLG tetrapeptide in lysosomes, precisely releasing active PROTAC. In vivo experiments demonstrated a CDK4/6 degradation efficiency for PSRNs 2.6-3.0 times higher than that of free PROTAC. In combination with α-PD-1 therapy, PSRNs significantly inhibited tumor growth in the CT26 CRC model and markedly prolonged survival of treated mice[124].
The combined response to pH and GSH also demonstrates excellent therapeutic outcomes in lung cancer. Guan et al. developed the RLDPB [cRGD/Lewis Lung Cancer (LLC) membrane/DS-PLGA/dBET6, RLDPB is a nano-PROTAC drug named by the authors] micelles nano-system utilizing disulfide-linked PLGA (DS-PLGA) carriers to encapsulate BRD4-targeting PROTAC (dBET6), with surface modification by LLC cell membranes and cRGD ligands. The acidic intracellular environment accelerates PLGA hydrolysis, while high GSH concentrations cleave disulfide bonds, resulting in dual-responsive carrier degradation. The dual response to pH (5.5) and high GSH levels (5-10 mM) significantly enhances dBET6 release, achieving an 80% release rate. RLDPB efficiently degrades BRD4 in LLC cells, outperforming free dBET6 and single-responsive carriers. Tumor tissue drug enrichment is substantially improved, with fluorescence intensity 10.2 times greater than surrounding normal muscle[125]. Zhang et al. further expanded this concept by targeting TAMs with the CREATE (CRV-LLC membrane/DS-PLGA/dBET6, CREATE is a nano-PROTAC drug named by the authors) system, which is constructed by using a pH/GSH (glutathione)-responsive polymer (disulfide bond-linked poly(lactic-co-glycolic acid), DS-PLGA) to load BRD4-targeted PROTAC (dBET6)[126]. They achieved dual targeting via LLC cell membranes modified with CRV-Lamp2 fusion protein (CRV-LLCM). This modification allows homotypic targeting of lung cancer cells and specific binding to TAMs through the CRV ligand. The pH/GSH-responsive release simultaneously degrades BRD4 in both cell types, reshaping the tumor immune microenvironment. In orthotopic lung cancer models, CREATE significantly suppressed tumor growth, extended survival (90% of tumor-bearing mice survived beyond 18 days), and significantly reduced lung tumor weight compared with controls.
Combination of ROS with copper ion signals
The synergy between ROS and copper ions presents a novel strategy for cuproptosis-sensitizing PROTACs. Wang et al. developed a nano-system (CuS-MD@CS) based on PROTAC sensitized for cuproptosis. Targeting the elevated ROS levels and copper ion accumulation characteristic of lung cancer cells, the researchers loaded the MDM2-targeting PROTAC (MD-224) into copper sulfide (CuS) nanoparticles capable of releasing copper ions. Endogenous ROS in the TME triggers nanoparticle dissociation, releasing both PROTAC and copper ions (Cu2+) to create a dual “ROS - copper ion” signal. On one hand, PROTAC degrades MDM2, upregulating p53 to suppress glycolysis and cell cycle progression. On the other hand, ROS and copper ions jointly promote lipid peroxidation, enhancing cuproptosis. In vitro experiments demonstrated that this system significantly increased the apoptosis rate of A549 cells compared to free PROTAC. In vivo, the lung cancer model showed considerable suppression of tumor growth and marked reduction in tumor volume relative to controls[127].
Endogenous-exogenous combined activation systems
The endogenous - exogenous co-activated nano-TPD system integrates a nanocarrier core with dual activation modules: “endogenous TME signals” (e.g., pH, enzyme, hypoxia, or ROS/GSH) and “externally controlled exogenous signals” (e.g., light, ultrasound, or magnetic fields)[128]. This technology triggers TPD via cascaded synergy between these two signal types. Its core advantage is “dual-signal-dependent precise activation”, exogenous signals provide controlled spatial and temporal initiation, while endogenous signals ensure specificity and amplify degradation effects within target regions. Requiring simultaneous activation by multiple signals improves targeting specificity, reducing off-target effects associated with single-signal fluctuations. By integrating endogenous targeting and exogenous control, the system achieves progressive “tumor localization → on-demand degradation”, adapting to complex pathological environments. This approach addresses TME heterogeneity by responding to multiple signals, covering broader pathological conditions. It also significantly reduces PROTAC exposure in healthy tissues, representing one of the most clinically promising systems currently available[129].
Existing systems generally achieve synergy via two cascade modes: “exogenous signal localization → endogenous signal activation” or “exogenous signal triggering → endogenous signal amplification”. Relevant literature is classified by the type of exogenous signal applied.
Combination of light and enzyme signals
Systems centered on exogenous laser-triggered ROS generation and endogenous CTSB activation achieve tumor-specific protein degradation and immune activation. Park et al. designed a nano-PROTAC termed NanoTAC, self-assembled from hexokinase II (HK2)-targeting PROTAC and photosensitizer verteporfin (VPF) linked via a CatB-sensitive peptide (KRR)[130]. Upon laser irradiation, VPF generates ROS, causing tumor cell pyroptosis and release of tumor-associated antigens (TAAs) to stimulate immune responses. Concurrently, tumor-overexpressed CatB cleaves the KRR peptide, releasing PROTAC to degrade HK2, thereby suppressing glycolysis and mitochondrial respiration. This dual-signal synergy enabled significantly superior HK2 degradation compared to free PROTAC and traditional HK2 inhibitors, achieving a 60% complete tumor regression rate in the 4T1 triple-negative breast cancer model.
Choi et al. developed a light-triggered PROTAC nanoassembly (LPN) via self-assembly of an indoleamine 2,3-dioxygenase (IDO)-targeting PROTAC, CatB-sensitive peptide (KRR), and VPF. Under laser irradiation, VPF-produced ROS induce ICD. Endogenous CatB cleaves the peptide, releasing active PROTAC, which degrades IDO and reverses tryptophan metabolism-mediated immunosuppression. This dual-signal synergy achieved a 100% complete tumor regression rate in a CT26 colon cancer model, effectively preventing metastasis and recurrence[131].
Zhang et al. developed a semiconducting polymer-based nano-PROTAC system (SPNpro)[132]. This system contains a semiconducting polymer core linked to an IDO-targeting PROTAC fragment (IPP), comprising IDO inhibitor (NLG919) and an E3 ubiquitin ligase VHL-binding peptide via a CatB-sensitive peptide. SPNpro exhibits both phototherapy and enzyme-activated protein degradation functions. After passive tumor accumulation via EPR effect, SPNpro generates 1O2 under NIR irradiation. On one hand, this directly kills tumor cells and triggers ICD, releasing TAAs to activate DCs and effector T cells. On the other hand, tumor-expressed CatB cleaves the linker, releasing PROTAC to continuously degrade immunosuppressive IDO via the UPS. Blocking the conversion of tryptophan (Trp) to kynurenine (Kyn) alleviates immune suppression. Experiments showed effective inhibition of primary tumor growth and distant metastasis in the 4T1 breast cancer model, significant enhancement of T-cell infiltration, and no notable toxicity to major organs.
Combination of light and hypoxia signals
The combination of exogenous laser-induced hypoxia and endogenous tumor hypoxia-responsive PROTAC creates a positive feedback loop to enhance protein degradation efficiency. Fu et al. developed a hypoxia-responsive PROTAC prodrug (AZO-PRO), linking BRD4-targeting PROTAC with the photosensitizer methylene blue (MB) through an azo bond[133]. Under exogenous irradiation (660 nm laser), MB-mediated PDT consumes oxygen in the tumor, increasing the hypoxic environment. Overexpressed azoreductases (e.g., NQO1) in this endogenous hypoxic environment subsequently cleave the azo bond, releasing active PROTAC and MB. Released PROTAC degrades BRD4, and MB further produces ROS to induce apoptosis, thus forming a “laser-enhanced hypoxia → hypoxia-activated PROTAC” positive feedback loop. This dual-signal synergy achieved superior BRD4 degradation compared to free PROTAC or hypoxia-only activated groups. In the MCF-7 tumor model, tumor growth was significantly suppressed, with tumor volume increasing only 1.51-fold, substantially lower than the 17.39-fold increase observed in the PBS control group.
Combination of light and pH signals
This approach combines exogenous laser-induced photothermal/PDT and endogenous acidic TME responsive PROTAC release, enhancing protein degradation and immune activation simultaneously. Li et al. developed a nano-PROTAC (ARV@PEG-ICG) by loading BRD4-targeting PROTAC (ARV-825) into PEG-modified indocyanine green (PEG-ICG) via nanoprecipitation. ARV-825 serves as the BRD4 degrader, while PEG-ICG acts as a photosensitizer. Under exogenous irradiation (808 nm laser), PEG-ICG generates ROS and photothermal effects, inducing tumor apoptosis and ICD. Endogenous acidic conditions (pH 5.5) trigger carrier depolymerization, releasing ARV-825 to degrade BRD4 and enhance phototherapy by inhibiting inducible nitric oxide synthase (iNOS). This dual-signal synergy achieved an 85% tumor inhibition rate in the 4T1 tumor model and stimulated systemic immune responses to suppress distant tumor growth[134].
Multi-signal combination of light, GSH, and pH
This system integrates exogenous laser-triggered carrier disassembly with endogenous TME GSH and pH signal-responsive PROTAC activation. It addresses TME-mediated inhibitory effects on protein degradation and cuproptosis by consuming GSH and regulating metabolic reprogramming. Lu et al. developed a nano-platform (HSA@PPa-JQ1) by encapsulating a photodegradation-targeting chimera (PDTAC) molecule (PPa-JQ1) within human serum albumin (HSA) carriers. PPa-JQ1 consists of BRD4-targeting ligand JQ1-acid covalently linked to the photosensitizer pyropheophorbide-a (PPa) via a linker. Exogenous irradiation (660 nm laser) enables PPa to produce ROS, causing tumor cell apoptosis. Meanwhile, the endogenous high GSH concentration and acidic environment (pH 5.5) trigger carrier depolymerization, releasing PPa-JQ1. Released PPa-JQ1 specifically targets and binds BRD4 through JQ1-acid, mediating BRD4 degradation via ROS generation. The dual-signal synergy achieved a 90% BRD4 degradation rate in the B16 melanoma model, significantly suppressing tumor growth, prolonging mouse survival, and demonstrating excellent systemic biocompatibility[135].
Combination of light and enzyme/hypoxia/pH signals
A positive feedback loop, integrating exogenous laser-triggered ROS and endogenous signals (MMP-2, pH, and hypoxia), addresses TME heterogeneity. Gao et al. developed a nanoplatform (PGDAT@N) co-loading a ROS-responsive PROTAC prodrug (ARV771-TK) and a hypoxia-responsive PROTAC prodrug (ARV771-Nb). Under exogenous 671 nm laser irradiation, the photosensitizer PPa generates ROS, cleaving the thioketal (TK) linker to release ARV771, which degrades BRD4 in normoxic tumor regions. Endogenously, overexpressed MMP-2 cleaves the GPLGLAG heptapeptide linker on the nanoparticle surface, removing the PEG corona to enhance tumor penetration. The acidic TME (pH < 6.2) protonates 2-(diisopropylamino)ethyl methacrylate (DPA) groups, causing nanoparticle dissociation and restoring PPa photoactivity. Concurrently, nitroreductase (NTR) overexpressed in hypoxic regions activates ARV771-Nb, degrading BRD4 in hypoxic cancer stem-like cells (CSCs). This multi-signal integration achieves comprehensive degradation of both normoxic and hypoxic tumor cells, effectively addressing tumor heterogeneity. In the MDA-MB-231 tumor model, the tumor inhibition rate reached 95%, with complete tumor regression in 5 of 6 mice[136].
Combination of ultrasonic and ROS signals
This approach integrates exogenous ultrasound-mediated deep tissue penetration with endogenous ROS activation to achieve targeted degradation in deep tumors. Liu et al. designed an ultrasound-ROS dual-responsive nanoparticle [NP(Ce6+PRO)] co-assembled from polyethylene glycol-b-polycaprolactone (PEG-b-PCL), sonosensitizer-conjugated polymer (Ce6-PCL), and ROS-responsive PROTAC conjugate (TK-PROTAC-PCL), loading a BRD4-targeted PROTAC precursor protected by a thioketal (TK) linker. Under exogenous ultrasound (1.0 MHz) irradiation of deep pancreatic tumors, nanoparticles dissociate locally. Ultrasound-induced ROS generation by Ce6 (endogenous signal) cleaves the TK linker, activating the PROTAC precursor. Ultrasound provides dual functions of deep-tissue penetration and local activation, overcoming traditional photoactivated PROTAC limitations. ROS ensures intracellular PROTAC activation specifically within tumors. Dual-signal synergy increased the PROTAC concentration in large pancreatic tumors by 3.09-fold compared to laser treatment alone, achieving a 68.5% BRD4 degradation rate and significantly inhibiting tumor growth[137].
Combination of magnetic and pH signals
Exogenous magnetic-field guidance combined with inflammation-specific targeting by M1 macrophage-derived exosome membranes enables selective nano-TPD accumulation in inflammatory regions associated with acute lung injury (ALI). Chen et al. constructed a nano-PROTAC (EMPLANT) using poly(lactic-co-glycolic acid) (PLGA) to encapsulate magnetic nanoparticles and TRIM24-targeted PROTAC (dTRIM24), coated with exosome membranes from M1 macrophages. An external magnetic field guides nano-TPD to pulmonary inflammatory regions in ALI mice. The endogenous acidic lysosomal environment (pH 5.0) triggers carrier release, liberating dTRIM24. Released dTRIM24 degrades TRIM24 via the UPS, upregulating STAT6 to promote macrophage polarization from M1 to M2 phenotype. The dual-signal synergy achieved a 90% TRIM24 degradation rate in lung tissues and increased mouse survival to nearly 100%[138].
In summary, multi-conditionally responsive nano-TPD systems represent a rational evolution of targeted protein degradation platforms, shifting the design paradigm from single-trigger activation toward sequential response or multiple stimulus response. By integrating endogenous pathological cues with complementary endogenous or exogenous signals, these systems strike a balance between biological specificity and external controllability. Such combinatorial activation strategies not only minimize unintended protein degradation but also provide a versatile framework for adapting nano-TPD platforms to diverse disease contexts and clinical constraints. As material engineering, stimulus delivery technologies, and PROTAC design continue to advance, multi-condition-responsive nano-TPD systems are expected to play a pivotal role in bridging the gap between mechanistic innovation and translational applicability.
PERFORMANCE COMPARISON AND KEY CHALLENGES
Performance comparison
Different nano-TPD systems vary substantially in core performance metrics, nano-TPD platforms should be benchmarked as a multi-objective trade-off[139]. In endogenous initiation, activation is “device-free” and typically shows favorable biocompatibility, yet the activation threshold, kinetics, and effective dose are tightly coupled to the spatially heterogeneous TME, which can yield patient-to-patient variability and incomplete degradation in poorly perfused or “cold” lesions. This dependence also complicates cross-study comparability because the same formulation may behave differently across models with distinct microenvironmental distributions[16].
In contrast, exogenous initiation decouples activation from TME variability by using externally applied signals that provide remote timing control and high spatiotemporal precision, thereby narrowing systemic exposure windows and potentially reducing off-target toxicity. However, these gains introduce new boundary conditions: tissue penetration depth and focal volume (notably for light), dependence on specialized equipment/clinical workflow, and the possibility of collateral activation when the stimulus field cannot be perfectly confined[140-142].
Multi-condition initiation partially reconciles the above by implementing logic-gated degradation (AND/sequential gating): endogenous cues act as a biological “permission” signal, while an exogenous input provides temporal/spatial “authorization”. Such architectures can enlarge the safety margin for targets with narrow therapeutic windows by requiring multiple conditions before degradation occurs. Nevertheless, adding triggers and functional modules typically increases synthetic complexity, quality control burden, and batch-to-batch reproducibility risk, which may dominate translational feasibility[136].
Finally, self-assembled nano-TPD systems reshape the performance profile by offering high apparent loading/modularity and improved intracellular delivery; yet their supramolecular heterogeneity, in vivo remodeling, and stimulus-independent leakage can confound mechanistic attribution. Therefore, future benchmarking should report not only tumor inhibition but also quantitative degradation kinetics (extent, onset, duration, reversibility) under clinically relevant conditions, alongside manufacturability and stability metrics[143] [Table 3].
Performance comparison of different types of conditionally starting nano-targeted degradation systems
| Activation type | Endogenous | Self-assembled endogenous | Exogenous | Self-assembled exogenous | Dual endogenous signal combined | Endogenous & exogenous signal combined |
| Targeting precision | Medium/high | Medium/high | High/very high | High/very high | Very high | Very high |
| Response rate | Tens of minutes to hours | Tens of minutes to hours | Seconds to minutes | Seconds to minutes | Hours | Slow enrichment, fast unlocking |
| Drug loading | Medium | High | Medium | High | Medium | Medium/high |
| Synthetic complexity | Low/medium | Medium | Medium | Medium/high | High | Very high |
| Tissue penetration depth | Inapplicability | Inapplicability | Signal limitation | Signal limitation | Inapplicability | Exogenous signal-dependent |
| External device requirement | No | No | Yes | Yes | No | Yes |
| Estimated cost | Low | Medium | Medium/high | Medium/high | Medium | High |
| Biocompatibility | Excellent | Excellent | Good/excellent | Good/excellent | Excellent | Good/excellent |
| Spatiotemporal controllability | Relying on microenvironment | Relying on microenvironment | Controllable by external signals | Controllable by external signals | Dependent on multiple microenvironmental signals | Two-level regulation |
| Application scenarios | Solid tumors with significant microenvironment abnormalities, chronic inflammation | Tumors requiring long-term administration, autoimmune diseases | Superficial & focusable lesions, postoperative residual tumors | High-precision treatment scenarios, multi-lesion synergistic therapy | Complex tumors with multiple microenvironment abnormalities | Central nervous system disease, critical organ disease |
Key challenges
Although nano-TPD systems have made considerable progress in basic research, several significant challenges remain for clinical translation[144]. These include balancing microenvironmental heterogeneity with individualized matching, ensuring equilibrium between carrier stability and response sensitivity, overcoming barriers in industrial-scale synthesis and quality control, and addressing issues related to pathway coordination and safety evaluation. Carrier fate (ADME), long-term toxicity, immunogenicity, and combination-product regulatory pathways should be explicitly addressed to strengthen the translational argument[143-146].
A major challenge for endogenous activation systems is microenvironmental heterogeneity, which complicates personalized matching. Microenvironment parameters (e.g., pH, enzyme activity, ROS levels, GSH concentration) differ substantially among patients, disease stages, and within lesions themselves. Designing a universal response threshold based on “average characteristics” to ensure efficacy and safety across cases is difficult[147]. Even in combined activation systems, precisely aligning patient-specific microenvironment features with exogenous stimuli remains challenging. Current methods for personalized assessment and customized design remain inadequate, highlighting the urgent need for precise, individualized treatment strategies to enhance therapeutic targeting and effectiveness[148]. Notably, in clinically approved nanomedicines, improving circulation alone does not guarantee improved tissue accessibility; rather, human - animal mismatches in distribution/clearance and tissue “gatekeeping” frequently dominate outcomes[149]. Therefore, nano-TPD designs that rely on microenvironment triggers should be coupled with biomarker-based stratification and Pharmacokinetics/Pharmacodynamics (PK/PD)-linked activation modeling to avoid under-activation (loss of efficacy) and over-activation (off-target toxicity) in heterogeneous clinical populations.
Balancing carrier stability with response sensitivity also represents a critical research challenge. Nano-TPD systems must maintain stability during systemic circulation yet rapidly respond at target sites. In self-assembled systems, factors such as plasma protein adsorption and blood shear forces can destabilize carriers, causing premature drug release or structural changes. In exogenously responsive systems, enhanced sensitivity can improve therapeutic outcomes but may compromise circulation stability[150-152]. Conversely, prioritizing stability may reduce response speed. Thus, identifying an optimal balance between these two aspects remains essential[153]. This balance should be evaluated within a full ADME framework - where the key questions are “Where does the carrier go? How long does it persist? How does it transform in vivo? How is it cleared?” A Nature Reviews Bioengineering review emphasizes that controlling blood circulation, biodistribution, tissue accessibility, and clearance is central to nanomedicine translation[149]. For nano-TPD, such ADME uncertainty directly affects degrader exposure, intracellular delivery, and ultimately degradation kinetics, making it insufficient to report only in vitro responsiveness or short-term tumor accumulation.
Safety concerns, such as long-term residues from inorganic nanocarriers and immunogenicity of orthogonal enzymes, lack comprehensive long-term toxicology data, undermining assurances of biosafety[154-156]. Clinically approved liposomal products (e.g., Doxil) demonstrate that nanomedicines can be successful, but also highlight that release testing, stability, and bioequivalence-sensitive quality attributes can be rate-limiting for regulatory review and lifecycle management[157]. FDA has specifically developed science and guidance around nanomaterial-containing products and liposome drug products, reflecting the regulatory focus on Chemistry, Manufacturing, and Controls (CMC), human PK/Bioequivalence (BE), labeling, and in vitro release testing for complex nanosystems[158]. Immunologically, PEGylated nanocarriers can trigger anti-PEG antibodies, leading to the accelerated blood clearance (ABC) phenomenon upon repeat dosing, reducing efficacy and altering safety[159]. In addition, liposomes and other nanoparticles may cause complement activation-related pseudoallergy (CARPA), an unpredictable hypersensitivity risk that has been discussed for decades and remains clinically relevant for reactogenic nanocarriers[160,161]. These well-documented phenomena imply that nano-TPD platforms - especially those intended for repeated administration - should incorporate immunotoxicity endpoints (complement activation, cytokine release, anti-carrier antibodies, ABC risk) early in development rather than treating them as late-stage toxicology add-ons[162].
For TPD itself, safety is not only material-driven: off-target degradation and system-level consequences of degrading proteins require proteomics-enabled selectivity/safety profiling to reduce unintended toxicity[163]. Thus, long-term biosafety packages should integrate both carrier-centric (retention/accumulation, chronic inflammation/fibrosis) and mechanism-centric (degradome/off-target) assessments.
Industrialization barriers in synthesis and quality control significantly hinder the clinical application of nano-TPD systems. Self-assembled and co-activated systems typically integrate multiple functional components (e.g., sensitive linkers, targeting ligands, responsive modules, TPD loading sites), resulting in complex structures, lengthy synthesis routes, difficulty in intermediate purification, and poor reproducibility between batches. During large-scale production, these complexities not only increase synthesis difficulties but also complicate establishing reliable quality control standards[164]. Furthermore, accurately characterizing nano-TPD behavior in vivo requires simultaneous monitoring of multiple processes, including carrier distribution, structural changes, TPD release, and target protein degradation. Current characterization methods lack real-time, in situ evaluation capabilities, limiting clinical applicability[165]. Regulators expect sponsors to define critical quality attributes (CQAs) and link them to performance. FDA guidance on drug products containing nanomaterials outlines development expectations, while liposome-specific guidance highlights CMC and human PK considerations; FDA’s continued work on nano-size complex product in vitro release testing underscores how release/transformations become pivotal for complex nanoformulations. Therefore, nano-TPD manufacturing should move toward QbD-driven process control, orthogonal characterization (size distribution, morphology, surface chemistry/ligand density, encapsulation vs. free drug, stability, release/activation kinetics), and clinically relevant comparability plans[166].
Finally, coordinating treatment pathways and ensuring safety in clinical translation are critical. Exogenous and co-activated systems typically require specialized equipment and professional operation, complicating therapy coordination[167,168]. Dosing schedules, pre-activation windows, and external stimulus parameters increase operational complexity and patient compliance challenges[167]. Regulatory evaluation criteria for “nanomedicine + external device” co-activation models remain incomplete, delaying clinical translation. FDA defines “combination products” (drug/device, biologic/device, etc.) and provides cGMP and premarket pathway principles for such products, underscoring that integrated drug - device development must address not only pharmacology but also device controls, quality systems, and review coordination[169]. Hence, nano-TPD co-activation platforms should pre-emptively plan for combination-product requirements: standardized stimulus parameters, operator-independent workflows, device validation, and integrated risk management.
Overall, advancing nano-TPD systems clinically requires technological breakthroughs, rigorous safety assessments, scalable manufacturing with defined CQAs, and standardized clinical/regulatory protocols to ensure feasibility and safety[170,171].
PROSPECTS
Performance comparisons show that different systems prioritize varying factors, such as targeting precision, response rates, or drug-loading ratios, necessitating tailored designs based on specific clinical indications. Importantly, future development of nano-TPD systems should move beyond conceptual optimization toward clinically translatable design principles[172]. Based on the performance analysis and key challenges discussed above, translation success will likely depend on three coordinated dimensions: patient stratification and biomarker-guided activation, predictable in vivo fate and safety profiling, and manufacturability under defined regulatory frameworks.
Future research should emphasize personalized treatment strategies and technological optimization to advance nano-TPD applications in precision medicine[173]. Individualized design strategies will become critical, especially through integrating actual microenvironment parameters from patient lesions, enabling customized nano-TPD systems[174]. Accurately understanding patient-specific tumor or lesion characteristics will support developing personalized nanosystems that match biomarkers and therapeutic needs, significantly enhancing targeting accuracy and treatment efficacy[175,176]. Building on this foundation, to further boost translational feasibility, a more clinically actionable strategy can prioritize biomarker-defined patient subgroups and establish PK/PD-linked activation windows - ensuring that the responsiveness of nano-TPD systems aligns with quantifiable clinical parameters. This approach is expected to mitigate heterogeneity-induced variability and improve the reproducibility of therapeutic outcomes across diverse patient populations[149].
Additionally, increasing complexity in disease diagnosis and treatment emphasizes the need to optimize intelligent response mechanisms. Future studies should focus on advanced logic-gate response systems (e.g., “AND/OR/NOT” composite logic) that intelligently trigger responses based on multiple lesion features, substantially enhancing lesion identification and therapeutic responsiveness. These intelligent systems can rapidly respond to dynamic lesion changes, achieving efficient protein degradation and disease control[177-179]. Of course,system complexity must be balanced with manufacturability and regulatory feasibility. Overengineered logic-gate systems may face significant scale-up, quality control, and comparability challenges under current regulatory frameworks for nanomaterials and combination products. Thus, future intelligent designs should integrate QbD principles from early-stage development to ensure defined CQAs and scalable production pathways.
In technological advancements, innovations in multimodal characterization techniques will become essential[180]. Developing multimodal imaging technologies capable of real-time, in situ tracking of nano-TPD assembly, activation, and degradation processes will allow dynamic evaluation of key therapeutic steps and drug targeting efficiency[181-183]. More importantly, clinical translation requires standardized ADME and immunogenicity evaluation frameworks. Lessons from approved liposomal drugs and PEGylated nanoparticles indicate that ABC, CARPA, and long-term organ accumulation must be systematically assessed, particularly for repeated dosing regimens[158,159].
Furthermore, enhancing biosafety remains central. Researchers should develop fully biodegradable, low-immunogenicity nanocarriers and conduct comprehensive long-term safety evaluations[184]. Addressing potential risks associated with nanocarrier accumulation will ensure sustainable clinical use, particularly vital for patient safety during prolonged treatment[185]. Accelerating clinical translation requires simplifying system design and manufacturing, establishing standardized quality control methods, and developing effective clinical trial pathways[186].In this context, early integration of regulatory science - such as defining CQAs, implementing in vitro release testing (IVRT) strategies, and preparing for potential drug - device combination product classification - may substantially shorten development timelines.
Integrating artificial intelligence, particularly for data processing, predictive modeling, and real-time monitoring, will greatly enhance system intelligence and precision[187,188]. Artificial intelligence technology can optimize therapeutic regimens, predict drug responses, and adjust therapies in real time, accelerating clinical translation while improving therapeutic outcomes and safety[189-191].
Ultimately, the future of nano-TPD may not lie in maximal structural complexity, but in achieving a clinically predictable balance between targeting precision, safety margin, manufacturability, and regulatory clarity. Bridging mechanistic innovation with translational discipline will be the decisive step in transforming nano-TPD from a promising research platform into a viable therapeutic modality for malignancies and other refractory diseases.
CONCLUSIONS
This review systematically summarizes the design principles and activation strategies of nano-TPD by classifying them according to their activation triggers. By integrating nanotechnology with TPD approaches, nano-TPD systems offer diverse strategies to overcome the delivery limitations of conventional TPD technologies. Based on their activation mechanisms, nano-TPD platforms can be broadly categorized into endogenous activation, exogenous activation, and combined activation systems, each providing distinct advantages in targeting capability, spatiotemporal control, or drug-loading efficiency. However, these systems also face specific challenges, including microenvironment heterogeneity, reliance on external devices, stability issues, and design complexity, indicating that activation strategies must be rationally selected according to clinical application requirements.
Future development of nano-TPD technology should prioritize clinical translation, with focused efforts on personalized design, intelligent responsiveness, technological innovation, and safety and regulatory compliance, to enable the transition from a research concept to a viable therapeutic modality.
DECLARATIONS
Acknowledgments
The authors sincerely thank the FigDraw platform for providing drawing support for this article.
Authors’ contributions
Conceptualization, methodology and writing of the draft: Liu, Z.; Zhang, G.
Validation: Yan, S.; Xiao, J.; Cao, Z.; Guo, X.; Liao, G.; Peng, R.; Liu, X.; Tan, Z.; Xie, Q.; Luo, W.
Supervision and funding acquisition: Xiao, X.; Qin, S.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Hunan Science Fund for Distinguished Young Scholars (No. 2025JJ20098); the National Natural Science Foundation of China (No. 82204250); High-level Talent Research Initiation Fund Project of Hunan University of Chinese Medicine (No. 0004010, 0003003002); the National Natural Science Foundation of China (No.82503028).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Cromm, P. M.; Crews, C. M. Targeted protein degradation: from chemical biology to drug discovery. Cell. Chem. Biol. 2017, 24, 1181-90.
2. Farley, K.; Bhattacharya, S.; Cleland, J.; Chandran, P.; Wu, J. The targeted protein degradation landscape. Nat. Rev. Drug. Discov. 2025, 24, 164-5.
3. Zhang, G.; Yan, S.; Liu, Y.; Du, Z.; Min, Q.; Qin, S. PROTACs coupled with oligonucleotides to tackle the undruggable. Bioanalysis 2025, 17, 261-76.
4. Zhang, C.; Liu, Y.; Li, G.; et al. Targeting the undruggables-the power of protein degraders. Sci. Bull. 2024, 69, 1776-97.
5. Yan, S.; Zhang, G.; Luo, W.; et al. PROTAC technology: From drug development to probe technology for target deconvolution. Eur. J. Med. Chem. 2024, 276, 116725.
6. Yoon, H.; Rutter, J. C.; Li, Y. D.; Ebert, B. L. Induced protein degradation for therapeutics: past, present, and future. J. Clin. Invest. 2024, 134, e175265.
7. Kelm, J. M.; Pandey, D. S.; Malin, E.; et al. PROTAC'ing oncoproteins: targeted protein degradation for cancer therapy. Mol. Cancer. 2023, 22, 62.
8. Qin, S.; Xiao, X. Key advances and application prospects of PROTAC technologies in the next 5 years. Future. Med. Chem. 2025, 17, 987-9.
9. Kim, J.; Byun, I.; Kim, D. Y.; Joh, H.; Kim, H. J.; Lee, M. J. Targeted protein degradation directly engaging lysosomes or proteasomes. Chem. Soc. Rev. 2024, 53, 3253-72.
10. Zhou, Z.; Liang, J.; Cheng, B.; et al. Targeted degradation technologies utilizing autophagy. Int. J. Mol. Sci. 2025, 26, 6576.
11. Xie, H.; Zhang, C. Potential of the nanoplatform and PROTAC interface to achieve targeted protein degradation through the ubiquitin-proteasome system. Eur. J. Med. Chem. 2024, 267, 116168.
12. Gonçalves, A. L.; Cunha, P. M.; da Silva Lima, A.; Dos Santos, J. C.; Segato, F. Production of recombinant lytic polysaccharide monooxygenases and evaluation effect of its addition into Aspergillus fumigatus var. niveus cocktail for sugarcane bagasse saccharification. Biochim. Biophys. Acta. Proteins. Proteom. 2023, 1871, 140919.
13. Price, E.; Weinheimer, M.; Rivkin, A.; et al. Beyond rule of five and PROTACs in modern drug discovery: polarity reducers, chameleonicity, and the evolving physicochemical landscape. J. Med. Chem. 2024, 67, 5683-98.
14. Pike, A.; Williamson, B.; Harlfinger, S.; Martin, S.; McGinnity, D. F. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug. Discov. Today. 2020, 25, 1793-800.
15. Shi, J.; Wang, L.; Zeng, X.; et al. Precision-engineered PROTACs minimize off-tissue effects in cancer therapy. Front. Mol. Biosci. 2024, 11, 1505255.
16. Syahputra, E. W.; Lee, H.; Cho, H.; Park, H. J.; Park, K. S.; Hwang, D. PROTAC delivery strategies for overcoming physicochemical properties and physiological barriers in targeted protein degradation. Pharmaceutics 2025, 17, 501.
17. Toriki, E. S.; Papatzimas, J. W.; Nishikawa, K.; et al. Rational chemical design of molecular glue degraders. ACS. Cent. Sci. 2023, 9, 915-26.
18. Stanton, C.; Nutsch, K.; Bollong, M. J. Stressing out with electrophiles. Nat. Chem. Biol. 2025, 21, 612-3.
19. Zhang, X. J.; Chen, S.; Huang, K. X.; Le, W. D. Why should autophagic flux be assessed? Acta. Pharmacol. Sin. 2013, 34, 595-9.
20. Wu, Y.; Lin, B.; Lu, Y.; et al. Aptamer-LYTACs for targeted degradation of extracellular and membrane proteins. Angew. Chem. Int. Ed. 2023, 62, e202218106.
21. Mikitiuk, M.; Barczyński, J.; Bielski, P.; et al. IGF2 peptide-based LYTACs for targeted degradation of extracellular and transmembrane proteins. Molecules 2023, 28, 7519.
22. Mu, X.; Mu, C.; Wang, M.; Meng, C.; Liu, H.; Lu, H. Nanotechnology advances proteolysis targeting chimeras (PROTACs): transition from basic research to clinical application. Int. J. Nanomedicine. 2025, 20, 12177-98.
23. Peer, D.; Karp, J. M.; Hong, S.; Farokhzad, O. C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751-60.
24. Mitchell, M. J.; Billingsley, M. M.; Haley, R. M.; Wechsler, M. E.; Peppas, N. A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug. Discov. 2021, 20, 101-24.
25. Wilhelm, S.; Tavares, A. J.; Dai, Q.; et al. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014.
26. Rodriguez‐abetxuko, A.; Romero‐ben, E.; Ontoria, A.; et al. Engineered metal‐loaded biohybrids to promote the attachment and electron‐shuttling between enzymes and carbon electrodes. Adv. Funct. Mater. 2024, 34, 2400479.
27. Zhong, J.; Zhao, R.; Wang, Y.; Su, Y. X.; Lan, X. Nano-PROTACs: state of the art and perspectives. Nanoscale 2024, 16, 4378-91.
28. Kim, J.; Cho, H.; Lim, D. K.; Joo, M. K.; Kim, K. Perspectives for improving the tumor targeting of nanomedicine via the EPR Effect in clinical tumors. Int. J. Mol. Sci. 2023, 24, 10082.
29. Sun, J.; Gu, M.; Peng, L.; et al. A Self-assembled nano-molecular glue (Nano-mGlu) enables GSH/H2O2-triggered targeted protein degradation in cancer therapy. J. Am. Chem. Soc. 2025, 147, 372-83.
30. Xu, M.; Chen, J.; Wang, S.; et al. Autophagy-activating nanoautophagosome-tethering compounds for targeted protein degradation specifically in tumor cells. ACS. Macro. Lett. 2025, 14, 250-7.
31. Xing, Y.; Li, J.; Wang, L.; et al. A bifunctional lysosome-targeting chimera nanoplatform for tumor-selective protein degradation and enhanced cancer immunotherapy. Adv. Mater. 2025, 37, e2417942.
32. Zhong, G.; Chang, X.; Xie, W.; Zhou, X. Targeted protein degradation: advances in drug discovery and clinical practice. Signal. Transduct. Target. Ther. 2024, 9, 308.
33. Kashyap, D.; Milne, T.; Booth, M. DNA-programmable protein degradation: dynamic control of PROTAC activity via DNA hybridization and strand displacement. ChemRxiv. 2025.
34. Torres, J.; Valenzuela Oses, J. K.; Rabasco-Álvarez, A. M.; González-Rodríguez, M. L.; García, M. C. Innovations in cancer therapy: endogenous stimuli-responsive liposomes as advanced nanocarriers. Pharmaceutics 2025, 17, 245.
35. Thomas, R. G.; Surendran, S. P.; Jeong, Y. Y. Tumor microenvironment-stimuli responsive nanoparticles for anticancer therapy. Front. Mol. Biosci. 2020, 7, 610533.
37. Sahay, G.; Alakhova, D. Y.; Kabanov, A. V. Endocytosis of nanomedicines. J. Control. Release. 2010, 145, 182-95.
38. Li, L. M.; Xie, Y. P.; Qin, Y. R.; et al. Tumor microenvironment‐responsive drug self‐delivery systems to treat cancer and overcome MDR. Rare. Metals. 2024, 44, 1-33.
39. Liu, Y.; Si, L.; Jiang, Y.; et al. Design of pH-responsive nanomaterials based on the tumor microenvironment. Int. J. Nanomedicine. 2025, 20, 705-21.
40. Pei, D.; Buyanova, M. Overcoming endosomal entrapment in drug delivery. Bioconjugate. Chem. 2019, 30, 273-83.
41. Varkouhi, A. K.; Scholte, M.; Storm, G.; Haisma, H. J. Endosomal escape pathways for delivery of biologicals. J. Control. Release. 2011, 151, 220-8.
42. Huh, K. M.; Kang, H. C.; Lee, Y. J.; Bae, Y. H. pH-sensitive polymers for drug delivery. Macromol. Res. 2012, 20, 224-33.
43. Akinc, A.; Thomas, M.; Klibanov, A. M.; Langer, R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene. Med. 2005, 7, 657-63.
44. Shin, Y.; Husni, P.; Kang, K.; et al. Recent advances in pH- or/and photo-responsive nanovehicles. Pharmaceutics 2021, 13, 725.
45. Kanamala, M.; Wilson, W. R.; Yang, M.; Palmer, B. D.; Wu, Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: a review. Biomaterials 2016, 85, 152-67.
46. Huang, L.; Sun, X.; Zuo, Q.; et al. A pH-responsive PROTAC-based nanosystem triggers tumor-specific ferroptosis to construct in situ tumor vaccines. Mater. Today. Bio. 2025, 31, 101523.
47. Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941-51.
48. Du, J.; Lane, L. A.; Nie, S. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J. Control. Release. 2015, 219, 205-14.
49. Cui, M.; Zhang, D.; Zheng, X.; et al. Intelligent modular DNA lysosome-targeting chimera nanodevice for precision tumor therapy. J. Am. Chem. Soc. 2024, 146, 29609-20.
50. Min, H.; Sun, M. C.; Hu, K.; et al. Ionizable nano-PROTAC overcomes endosomal entrapment for enhanced LRG1 degradation and tumor suppression. Angew. Chem. Int. Ed. 2026, 65, e20235.
51. Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010, 141, 52-67.
52. Wang, H. X.; Yang, X. Z.; Sun, C. Y.; Mao, C. Q.; Zhu, Y. H.; Wang, J. Matrix metalloproteinase 2-responsive micelle for siRNA delivery. Biomaterials 2014, 35, 7622-34.
53. Pang, Q.; Xu, Z.; Sun, T.; et al. Strategic chemical synthesis and application of nanocarriers responsive to the tumor microenvironment. Nano. Today. 2024, 58, 102421.
54. Kim, D.; Yang, G.; Lim, C.; et al. Cancer specific CAIX-targeting supramolecular lysosome-targeting chimeras (Supra-LYTAC) for targeted protein degradation. Adv. Sci. 2025, 12, e2503134.
55. Kapalatiya, H.; Madav, Y.; Tambe, V. S.; Wairkar, S. Enzyme-responsive smart nanocarriers for targeted chemotherapy: an overview. Drug. Deliv. Transl. Res. 2022, 12, 1293-305.
56. Tong, F.; Wang, Y.; Xu, Y.; et al. MMP-2-triggered, mitochondria-targeted PROTAC-PDT therapy of breast cancer and brain metastases inhibition. Nat. Commun. 2024, 15, 10382.
57. Song, C.; Jiao, Z.; Hou, Z.; et al. Selective protein of interest degradation through the split-and-mix liposome proteolysis targeting chimera approach. J. Am. Chem. Soc. 2023, 145, 21860-70.
58. Yang, F.; Luo, Q.; Wang, Y.; et al. Targeted biomolecule regulation platform: a split-and-mix PROTAC approach. J. Am. Chem. Soc. 2023, 145, 7879-87.
59. Zhou, Z.; Ni, K.; Deng, H.; Chen, X. Dancing with reactive oxygen species generation and elimination in nanotheranostics for disease treatment. Adv. Drug. Delivery. Rev. 2020, 158, 73-90.
60. Yang, B.; Chen, Y.; Shi, J. Reactive oxygen species (ROS)-based nanomedicine. Chem. Rev. 2019, 119, 4881-985.
61. Zhao, C.; Chen, J.; Zhong, R.; Chen, D. S.; Shi, J.; Song, J. Oxidative-species-selective materials for diagnostic and therapeutic applications. Angew. Chem. Int. Ed. 2021, 60, 9804-27.
62. Ye, H.; Zhou, Y.; Liu, X.; et al. Recent advances on reactive oxygen species-responsive delivery and diagnosis system. Biomacromolecules 2019, 20, 2441-63.
63. Lee, M. H.; Yang, Z.; Lim, C. W.; et al. Disulfide-cleavage-triggered chemosensors and their biological applications. Chem. Rev. 2013, 113, 5071-109.
64. Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid. Med. Cell. Longev. 2019, 2019, 3150145.
65. Liu, H.; Ren, C.; Sun, R.; et al. Reactive oxygen species-responsive Pre-PROTAC for tumor-specific protein degradation. Chem. Commun. 2022, 58, 10072-5.
66. Bi, T.; Liang, P.; Zhao, Q.; et al. Targeted degradation of bromodomain-containing protein 4 enabled by reactive oxygen species-activatable NanoPROTACs as an efficient strategy to reverse liver fibrosis in chronic liver injury. J. Med. Chem. 2025, 68, 6328-38.
67. Yao, Q.; Wu, Z.; Li, J.; et al. Reactive oxygen species-instructed supramolecular assemblies enable bioorthogonally activatable protein degradation for pancreatic cancer. J. Am. Chem. Soc. 2025, 147, 18208-18.
68. Liu, H. J.; Chen, W.; Wu, G.; et al. Glutathione-scavenging nanoparticle-mediated PROTACs delivery for targeted protein degradation and amplified antitumor effects. Adv. Sci. 2023, 10, e2207439.
69. Lin, J. Y.; Wu, Y.; Liang, X. H.; et al. A self-assembling LYTAC mediates CTGF degradation and remodels inflammatory tumor microenvironment for triple-negative breast cancer therapy. Adv. Sci. 2025, 12, e2500311.
70. Saravanakumar, G.; Kim, J.; Kim, W. J. Reactive-oxygen-species-responsive drug delivery systems: promises and challenges. Adv. Sci. 2017, 4, 1600124.
72. Xie, F.; Wang, M.; Chen, Q.; et al. Endogenous stimuli-responsive nanoparticles for cancer therapy: from bench to bedside. Pharmacol. Res. 2022, 186, 106522.
73. Gao, J.; Yang, L.; Lei, S.; et al. Stimuli-activatable PROTACs for precise protein degradation and cancer therapy. Sci. Bull. 2023, 68, 1069-85.
74. Zhu, M.; Wang, X.; Zhou, Y.; et al. Breast tumor‐targeted drug delivery via polymer nanocarriers: endogenous and exogenous strategies. J. Appl. Polym. Sci. 2023, 140, e54227.
75. Wang, N.; Cong, W.; Zhu, Y.; et al. Spatiotemporally controlled protein degradation via NIR-activatable PROTAC platform. Sci. China. Chem. 2025, 68, 6621-7.
76. Sheng, J.; Ma, T.; Wu, Y.; Wang, M. Programmable PROTAC delivery for precise and spatiotemporal protein degradation. Chem. Commun. 2026, 62, 2414-27.
77. Hu, H.; Busa, P.; Zhao, Y.; Zhao, C. Externally triggered drug delivery systems. Smart. Mater. Med. 2024, 5, 386-408.
78. Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991-1003.
79. Liu, X.; He, F.; Liu, M. New opportunities of stimulus-responsive smart nanocarriers in cancer therapy. Nano. Mater. Sci. 2024.
80. Cheng, L.; Wang, C.; Feng, L.; Yang, K.; Liu, Z. Functional nanomaterials for phototherapies of cancer. Chem. Rev. 2014, 114, 10869-939.
81. Zhang, L.; Wang, D.; Yang, K.; et al. Mitochondria-targeted artificial “Nano-RBCs” for amplified synergistic cancer phototherapy by a single NIR irradiation. Adv. Sci. 2018, 5, 1800049.
82. Castano, A. P.; Mroz, P.; Hamblin, M. R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer. 2006, 6, 535-45.
83. Sun, B.; Bte Rahmat, J. N.; Zhang, Y. Advanced techniques for performing photodynamic therapy in deep-seated tissues. Biomaterials 2022, 291, 121875.
84. Li, J.; Pu, K. Semiconducting polymer nanomaterials as near-infrared photoactivatable protherapeutics for cancer. Acc. Chem. Res. 2020, 53, 752-62.
85. Mouret, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13765-70.
86. Zhan, J.; Li, X.; Mu, Y.; Yao, H.; Zhu, J. J.; Zhang, J. A photoactivatable upconverting nanodevice boosts the lysosomal escape of PROTAC degraders for enhanced combination therapy. Biomater. Sci. 2024, 12, 3686-99.
87. He, Q.; Zhou, L.; Yu, D.; et al. Near-infrared-activatable PROTAC nanocages for controllable target protein degradation and on-demand antitumor therapy. J. Med. Chem. 2023, 66, 10458-72.
88. Liu, J.; Chen, H.; Ma, L.; et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020, 6, eaay5154.
89. Wang, W.; Zhu, C.; Zhang, B.; Feng, Y.; Zhang, Y.; Li, J. Self-assembled nano-PROTAC enables near-infrared photodynamic proteolysis for cancer therapy. J. Am. Chem. Soc. 2023, 145, 16642-9.
90. Lu, X.; Wang, X.; Liao, H.; Sha, B.; Zhang, Y.; Li, J. Tandem-activatable PROTAC prodrug for tumor biomarker-driven near-infrared opto-proteolysis. Angew. Chem. Int. Ed. 2025, 64, e202510641.
91. Li, J.; Luo, Y.; Zeng, Z.; et al. Precision cancer sono-immunotherapy using deep-tissue activatable semiconducting polymer immunomodulatory nanoparticles. Nat. Commun. 2022, 13, 4032.
92. Huang, P.; Qian, X.; Chen, Y.; et al. Metalloporphyrin-encapsulated biodegradable nanosystems for highly efficient magnetic resonance imaging-guided sonodynamic cancer therapy. J. Am. Chem. Soc. 2017, 139, 1275-84.
93. Ouyang, J.; Tang, Z.; Farokhzad, N.; et al. Ultrasound mediated therapy: recent progress and challenges in nanoscience. Nano. Today. 2020, 35, 100949.
94. Xu, M.; Hu, Y.; Wu, J.; Liu, J.; Pu, K. Sonodynamic nano-LYTACs reverse tumor immunosuppressive microenvironment for cancer immunotherapy. J. Am. Chem. Soc. 2024, 146, 34669-80.
95. Zhang, Q.; Bao, C.; Cai, X.; et al. Sonodynamic therapy-assisted immunotherapy: a novel modality for cancer treatment. Cancer. Sci. 2018, 109, 1330-45.
96. Canavese, G.; Ancona, A.; Racca, L.; et al. Nanoparticle-assisted ultrasound: a special focus on sonodynamic therapy against cancer. Chem. Eng. J. 2018, 340, 155-72.
97. Fan, X.; Jiao, G.; Zhao, W.; Jin, P.; Li, X. Magnetic Fe3O4-graphene composites as targeted drug nanocarriers for pH-activated release. Nanoscale 2013, 5, 1143-52.
98. Najafi, N.; Kheirollah, J.; Majidi, H.; et al. Enhancing magnetic drug targeting efficiency through computational analysis: Investigating Particle-RBC interaction and non-newtonian blood behavior. J. Magn. Magn. Mater. 2024, 600, 172153.
99. Jiao, W.; Dai, L.; Yan, B.; Lyu, Y.; Fan, H.; Liu, X. Heating up the immune battle: magnetic hyperthermia against cancer. Fundam. Res. 2025, 5, 2401-5.
100. Lapusan, R.; Borlan, R.; Focsan, M. Advancing MRI with magnetic nanoparticles: a comprehensive review of translational research and clinical trials. Nanoscale. Adv. 2024, 6, 2234-59.
101. Lv, J.; Yue, R.; Liu, H.; et al. Enzyme-activated nanomaterials for MR imaging and tumor therapy. Coord. Chem. Rev. 2024, 510, 215842.
102. Salahpour Anarjan, F. Active targeting drug delivery nanocarriers: ligands. Nano-Struct. Nano-Objects. 2019, 19, 100370.
103. Chen, Z.; Kankala, R. K.; Long, L.; Xie, S.; Chen, A.; Zou, L. Current understanding of passive and active targeting nanomedicines to enhance tumor accumulation. Coord. Chem. Rev. 2023, 481, 215051.
104. Huang, L.; Zhang, Q.; Long, J.; Liu, Z.; Sun, X. Construction of novel magnetic systems for cancer immunotherapy via cancer-immunity cycle circuits. J. Control. Release. 2025, 378, 38-59.
105. Zhang, H.; Wang, J.; Han, R.; Sun, B.; Luo, C. Bioorthogonal chemistry-driven anticancer nanotherapeutics. Trends. Chem. 2023, 5, 697-710.
106. Keppel, P.; Hecko, S.; Mikula, H. Click-triggered bioorthogonal bond-cleavage reactions. Top. Curr. Chem. 2025, 383, 25.
107. Oliveira, B. L.; Guo, Z.; Bernardes, G. J. L. Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895-950.
108. Huang, J.; Yao, Z.; Li, B.; Ping, Y. Targeted delivery of PROTAC-based prodrug activated by bond-cleavage bioorthogonal chemistry for microneedle-assisted cancer therapy. J. Control. Release. 2023, 361, 270-9.
109. Sletten, E. M.; Bertozzi, C. R. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 2009, 48, 6974-98.
110. Blackman, M. L.; Royzen, M.; Fox, J. M. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc. 2008, 130, 13518-9.
111. Xie, S.; Zhu, J.; Peng, Y.; et al. In vivo self-assembly of PROTACs by bioorthogonal chemistry for precision cancer therapy. Angew. Chem. Int. Ed. 2025, 64, e202421713.
112. Gao, J.; Hou, B.; Zhu, Q.; et al. Engineered bioorthogonal POLY-PROTAC nanoparticles for tumour-specific protein degradation and precise cancer therapy. Nat. Commun. 2022, 13, 4318.
113. Sun, C.; Liu, S.; Lau, J. W.; Yang, H.; Chen, Y.; Xing, B. Inside Front Cover: Enzyme‐activated orthogonal proteolysis chimeras for tumor microenvironment‐responsive immunomodulation (Angew. Chem. Int. Ed. 22/2025). Angew. Chem. Int. Ed. 2025, 64, e202510013.
114. Swetha, K. L.; Roy, A. Tumor heterogeneity and nanoparticle-mediated tumor targeting: the importance of delivery system personalization. Drug. Deliv. Transl. Res. 2018, 8, 1508-26.
115. Li, S.; Zeng, Q.; Zhu, S.; et al. Engineering nano-PROTAC platforms: Self-assembly, stimuli-responsiveness, and targeted delivery for precision therapy. Chem. Eng. J. 2026, 531, 174085.
116. Zhou, T.; Gao, Y.; Ma, B.; et al. Multi-stimuli activated PROTAC prodrug for controlled protein degradation with enhanced therapeutic effects. Eur. J. Med. Chem. 2026, 306, 118571.
117. Shang, Y.; Zhu, L.; Xiao, Y.; et al. Logic gate activated lysosome targeting DNA nanodevice for controlled proteins degradation. Adv. Funct. Mater. 2023, 34, 2311722.
118. Weng, Z.; Yu, J.; Liao, Y.; Zhang, Y.; Chen, C.; Chen, W. Emerging strategies in smart nano-PROTAC for stimuli-responsive protein degradation and precision cancer therapy. Nano. Biomed. Eng. 2025, 17, 315-32.
119. Cai, J.; Chen, C.; Wang, J.; et al. PROTAC: a revolutionary technology propelling small molecule drugs into the next golden age. Front. Oncol. 2025, 15, 1676414.
120. Majumder, J.; Minko, T. Multifunctional and stimuli-responsive nanocarriers for targeted therapeutic delivery. Expert. Opin. Drug. Deliv. 2021, 18, 205-27.
121. Zhou, W.; Jia, Y.; Liu, Y.; Chen, Y.; Zhao, P. Tumor microenvironment-based stimuli-responsive nanoparticles for controlled release of drugs in cancer therapy. Pharmaceutics 2022, 14, 2346.
122. Hu, F.; Huang, J.; Bing, T.; et al. Stimulus-responsive copper complex nanoparticles induce cuproptosis for augmented cancer immunotherapy. Adv. Sci. 2024, 11, e2309388.
123. Poyia, F.; Neophytou, C. M.; Christodoulou, M. I.; Papageorgis, P. The role of tumor microenvironment in pancreatic cancer immunotherapy: current status and future perspectives. Int. J. Mol. Sci. 2024, 25, 9555.
124. Yang, L.; Yang, Y.; Zhang, J.; et al. Sequential responsive nano-PROTACs for precise intracellular delivery and enhanced degradation efficacy in colorectal cancer therapy. Signal. Transduct. Target. Ther. 2024, 9, 275.
125. Guan, X.; Xu, X.; Tao, Y.; et al. Dual targeting and bioresponsive nano-PROTAC induced precise and effective lung cancer therapy. J. Nanobiotechnol. 2024, 22, 692.
126. Zhang, H. T.; Peng, R.; Chen, S.; et al. Versatile nano-PROTAC-induced epigenetic reader degradation for efficient lung cancer therapy. Adv. Sci. 2022, 9, e2202039.
127. Wang, Y.; Yao, X.; Lu, Y.; et al. A PROTAC-based cuproptosis sensitizer in lung cancer therapy. Adv. Mater. 2025, 37, e2501435.
128. Mi, P. Stimuli-responsive nanocarriers for drug delivery, tumor imaging, therapy and theranostics. Theranostics 2020, 10, 4557-88.
129. Wang, L.; Li, J.; Zeng, X.; et al. Exogenous/endogenous stimuli-responsive antitumor prodrugs advance precision chemotherapy. Bioorg. Med. Chem. 2026, 132, 118485.
130. Park, B.; Choi, J.; Lee, J. H.; et al. Reprogramming of cancer metabolism via photoresponsive nano-PROTAC enhances pyroptosis-mediated immunotherapy. Signal. Transduct. Target. Ther. 2025, 10, 310.
131. Choi, J.; Park, B.; Park, J. Y.; et al. Light-Triggered PROTAC nanoassemblies for photodynamic IDO proteolysis in cancer immunotherapy. Adv. Mater. 2024, 36, e2405475.
132. Zhang, C.; Zeng, Z.; Cui, D.; et al. Semiconducting polymer nano-PROTACs for activatable photo-immunometabolic cancer therapy. Nat. Commun. 2021, 12, 2934.
133. Fu, Z.; Yang, C.; Yang, Y.; Pan, M.; Hou, H.; Li, J. Photodynamic enhancement of PROTAC prodrug activation in hypoxic tumors. Acta. Pharm. Sin. B. 2025, 15, 4945-60.
134. Li, Z.; Ren, G.; Wang, X.; et al. Tumor microenvironment responsive nano-PROTAC for BRD4 degradation enhanced cancer photo-immunotherapy. Biomaterials 2025, 322, 123387.
135. Lu, S.; Shi, Z.; Ding, C.; et al. BRD4-targeted photodegradation nanoplatform for light activatable melanoma therapy. Biomaterials 2025, 317, 123101.
136. Gao, J.; Jiang, X.; Lei, S.; et al. A region-confined PROTAC nanoplatform for spatiotemporally tunable protein degradation and enhanced cancer therapy. Nat. Commun. 2024, 15, 6608.
137. Liu, Y.; Wang, H.; Ding, M.; et al. Ultrasound-activated PROTAC prodrugs overcome immunosuppression to actuate efficient deep-tissue sono-immunotherapy in orthotopic pancreatic tumor mouse models. Nano. Lett. 2024, 24, 8741-51.
138. Chen, S.; Chen, E.; Su, J.; et al. Magnetically navigated nano-PROTAC ameliorates acute lung injury. J. Nanobiotechnol. 2025, 23, 622.
139. Dey, K.; Erwin, N.; Molina, N.; Chen, H.; He, M.; Zheng, G. Nano-TPD: Using nanoparticle-based systems to improve the delivery and therapeutic effect of targeted protein degraders. Med. Chem. Res. 2025, 34, 2205-27.
140. Zeng, S.; Zhang, H.; Shen, Z.; Huang, W. Photopharmacology of proteolysis-targeting chimeras: a new frontier for drug discovery. Front. Chem. 2021, 9, 639176.
141. Wang, Z. W.; Liu, Y.; Zhu, X. PhotoPROTACs: a novel biotechnology for cancer treatment. Trends. Cell. Biol. 2020, 30, 749-51.
142. Zhang, W.; Li, S.; Yuan, H.; Zhao, J. Advances in light-activated PROTACs and phototherapy-combined approaches for cancer treatment. Eur. J. Med. Chem. 2026, 302, 118310.
143. Hua, S.; de Matos, M. B. C.; Metselaar, J. M.; Storm, G. Current trends and challenges in the clinical translation of nanoparticulate nanomedicines: pathways for translational development and commercialization. Front. Pharmacol. 2018, 9, 790.
144. Baker, A. G.; Ho, A. P. T.; Itzhaki, L. S.; Fruk, L. Nanoparticle-mediated targeted protein degradation: an emerging therapeutics technology. Angew. Chem. Int. Ed. 2025, 64, e202503958.
145. Agrahari, V.; Agrahari, V. Facilitating the translation of nanomedicines to a clinical product: challenges and opportunities. Drug. Discov. Today. 2018, 23, 974-91.
146. Joyce, P.; Allen, C. J.; Alonso, M. J.; et al. A translational framework to DELIVER nanomedicines to the clinic. Nat. Nanotechnol. 2024, 19, 1597-611.
147. Sun, T.; Jiang, C. Stimuli-responsive drug delivery systems triggered by intracellular or subcellular microenvironments. Adv. Drug. Delivery. Rev. 2023, 196, 114773.
148. Hao, J.; Li, Y.; Huang, L.; et al. Smart nanoarchitectures for precision RNA delivery: harnessing endogenous and exogenous stimuli in cancer treatment. Theranostics 2025, 15, 7747-78.
149. Cabral, H.; Li, J.; Miyata, K.; Kataoka, K. Controlling the biodistribution and clearance of nanomedicines. Nat. Rev. Bioeng. 2023, 2, 214-32.
150. Fu, F.; Crespy, D.; Landfester, K.; Jiang, S. In situ characterization techniques of protein corona around nanomaterials. Chem. Soc. Rev. 2024, 53, 10827-51.
151. Ly, P.; Ly, K.; Phan, H.; Nguyen, H. H. T.; Duong, V.; Nguyen, H. V. Recent advances in surface decoration of nanoparticles in drug delivery. Front. Nanotechnol. 2024, 6, 1456939.
152. Zhang, Q.; Kuang, G.; Li, W.; Wang, J.; Ren, H.; Zhao, Y. Stimuli-responsive gene delivery nanocarriers for cancer therapy. NanoMicro. Lett. 2023, 15, 44.
153. Wan, J.; Huang, L.; Cheng, J.; Qi, H.; Jin, J.; Wang, H. Balancing the stability and drug activation in adaptive nanoparticles potentiates chemotherapy in multidrug-resistant cancer. Theranostics 2021, 11, 4137-54.
154. Chen, Z.; Li, H.; Bian, Y.; et al. Bioorthogonal catalytic patch. Nat. Nanotechnol. 2021, 16, 933-41.
155. Li, Y.; Vulpe, C.; Lammers, T.; Pallares, R. M. Assessing inorganic nanoparticle toxicity through omics approaches. Nanoscale 2024, 16, 15928-45.
156. Carter, P. J.; Quarmby, V. Immunogenicity risk assessment and mitigation for engineered antibody and protein therapeutics. Nat. Rev. Drug. Discov. 2024, 23, 898-913.
157. Mohamed, M.; Abu Lila, A. S.; Shimizu, T.; et al. PEGylated liposomes: immunological responses. Sci. Technol. Adv. Mater. 2019, 20, 710-24.
158. Miao, G.; He, Y.; Lai, K.; et al. Accelerated blood clearance of PEGylated nanoparticles induced by PEG-based pharmaceutical excipients. J. Control. Release. 2023, 363, 12-26.
159. Gaballa, S. A.; Shimizu, T.; Ando, H.; et al. Treatment-induced and pre-existing anti-peg antibodies: prevalence, clinical implications, and future perspectives. J. Pharm. Sci. 2024, 113, 555-78.
160. Szebeni, J. Complement activation-related pseudoallergy caused by liposomes, micellar carriers of intravenous drugs, and radiocontrast agents. Crit. Rev. Ther. Drug. Carrier. Syst. 2001, 18, 40.
161. Saxena, S.; Sharma, S.; Kumar, G.; Thakur, S. Unravelling the complexity of CARPA: a review of emerging advancements in therapeutic strategies. Arch. Dermatol. Res. 2025, 317, 439.
162. Szebeni, J. Complement activation-related pseudoallergy: a stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 2014, 61, 163-73.
163. Liu, X.; Zhang, Y.; Ward, L. D.; et al. A proteomic platform to identify off-target proteins associated with therapeutic modalities that induce protein degradation or gene silencing. Sci. Rep. 2021, 11, 15856.
164. Đorđević, S.; Gonzalez, M. M.; Conejos-Sánchez, I.; et al. Current hurdles to the translation of nanomedicines from bench to the clinic. Drug. Deliv. Transl. Res. 2022, 12, 500-25.
165. Wu, X.; Shu, Y.; Zheng, Y.; et al. Recent advances in nanomedicine: cutting-edge research on nano-PROTAC delivery systems for cancer therapy. Pharmaceutics 2025, 17, 1037.
166. Jayaraj, S.; Jiang, W.; Mudalige, T. An Automated capillary electrophoresis based method for drug release profiling of liposomal doxorubicin. J. Pharm. Sci. 2024, 113, 1088-93.
167. Overchuk, M.; Weersink, R. A.; Wilson, B. C.; Zheng, G. Photodynamic and photothermal therapies: synergy opportunities for nanomedicine. ACS. Nano. 2023, 17, 7979-8003.
168. Li, X.; Lovell, J. F.; Yoon, J.; Chen, X. Clinical development and potential of photothermal and photodynamic therapies for cancer. Nat. Rev. Clin. Oncol. 2020, 17, 657-74.
169. Reis, M. E.; Bettencourt, A.; Ribeiro, H. M. The regulatory challenges of innovative customized combination products. Front. Med. 2022, 9, 821094.
170. Li, H.; Shen, X.; Chu, Y.; Yuan, P.; Shuai, Q. Challenging and new opportunities for prodrug technology. Investig. New. Drugs. 2025, 43, 365-76.
171. Gupta, D. K.; Tiwari, A.; Yadav, Y.; Soni, P.; Joshi, M. Ensuring safety and efficacy in combination products: regulatory challenges and best practices. Front. Med. Technol. 2024, 6, 1377443.
172. Arenas-Moreira, M.; Ocaña, A.; Bravo, I.; Alonso-Moreno, C. PROTAC delivery systems: innovative approaches for cancer treatment. Biomed. Pharmacother. 2026, 194, 118892.
173. Wang, C.; Zhang, Y.; Chen, W.; Wu, Y.; Xing, D. New-generation advanced PROTACs as potential therapeutic agents in cancer therapy. Mol. Cancer. 2024, 23, 110.
174. Grasso, G.; Colella, F.; Forciniti, S.; et al. Fluorescent nano- and microparticles for sensing cellular microenvironment: past, present and future applications. Nanoscale. Adv. 2023, 5, 4311-36.
175. Sabit, H.; Pawlik, T. M.; Radwan, F.; et al. Precision nanomedicine: navigating the tumor microenvironment for enhanced cancer immunotherapy and targeted drug delivery. Mol. Cancer. 2025, 24, 160.
176. Mao, Y.; Xie, J.; Yang, F.; Luo, Y.; Du, J.; Xiang, H. Advances and prospects of precision nanomedicine in personalized tumor theranostics. Front. Cell. Dev. Biol. 2024, 12, 1514399.
177. Luo, C.; He, L.; Chen, F.; et al. Stimulus-responsive nanomaterials containing logic gates for biomedical applications. Cell. Rep. Phys. Sci. 2021, 2, 100350.
178. Tang, L.; Yang, Z.; Zhou, Z.; et al. A logic-gated modular nanovesicle enables programmable drug release for on-demand chemotherapy. Theranostics 2019, 9, 1358-68.
179. Jiang, C.; Zhao, Z.; East, A. K.; Bandyopadhyay, S.; Jiang, Z.; Chan, J. Logic-gated approach for targeted delivery and site-selective activation of photothermal agents in precision cancer treatment. Chem. Sci. 2025, 16, 5155-65.
180. Mishra, V.; Kumari, N.; Vyas, M.; Aljabali, A. A. A.; Chattaraj, A.; Mishra, Y. Advances in multimodal imaging techniques in nanomedicine: enhancing drug delivery precision. RSC. Adv. 2025, 15, 27187-209.
181. Saladino, G. M.; Mangarova, D. B.; Nernekli, K.; et al. Multimodal imaging approach to track theranostic nanoparticle accumulation in glioblastoma with magnetic resonance imaging and intravital microscopy. Nanoscale 2025, 17, 9986-95.
182. Ju, S.; Cho, H. Y. Biohybrid nanoparticle-based in situ monitoring of in vivo drug delivery. Biosensors 2023, 13, 1017.
183. Yang, Y.; Yue, S.; Qiao, Y.; et al. Activable multi-modal nanoprobes for imaging diagnosis and therapy of tumors. Front. Chem. 2020, 8, 572471.
184. Chen, Y.; Zhang, C.; Yu, Z.; Hu, K.; Zhou, T. Circumventing tumor immune evasion: coordinated immunotherapy via in situ tumor nanovaccines and local ablation. Chem. Eng. J. 2025, 523, 168636.
185. Parvin, N.; Joo, S. W.; Mandal, T. K. Biodegradable and stimuli-responsive nanomaterials for targeted drug delivery in autoimmune diseases. J. Funct. Biomater. 2025, 16, 24.
186. Zheng, C.; Li, M.; Ding, J. Challenges and opportunities of nanomedicines in clinical translation. Bio. Integration. 2021, 2, 57. https://bio-integration.org/10-15212-bioi-2021-0016/ (accessed 2026-04-24).
187. Han, Y.; Kim, D. H.; Pack, S. P. Nanomaterials in drug delivery: leveraging artificial intelligence and big data for predictive design. Int. J. Mol. Sci. 2025, 26, 11121.
188. Chakroborty, S.; Nath, N.; Sahoo, S.; et al. A review of emerging trends in nanomaterial-driven AI for biomedical applications. Nanoscale. Adv. 2025, 7, 3619-30.
189. DeRidder, L. B.; Hare, K. A.; Lopes, A.; et al. Closed-loop automated drug infusion regulator: a clinically translatable, closed-loop drug delivery system for personalized drug dosing. Med 2024, 5, 780-796.e10.
190. Paci, M. M.; Saha, T.; Djassemi, O.; et al. Smart closed-loop drug delivery systems. Nat. Rev. Bioeng. 2025, 3, 816-34.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].