



Theoretical investigation on the self-organized MXene heterostructures: interface, sliding and Li/Na ion storage application
 0
0 
Abstract
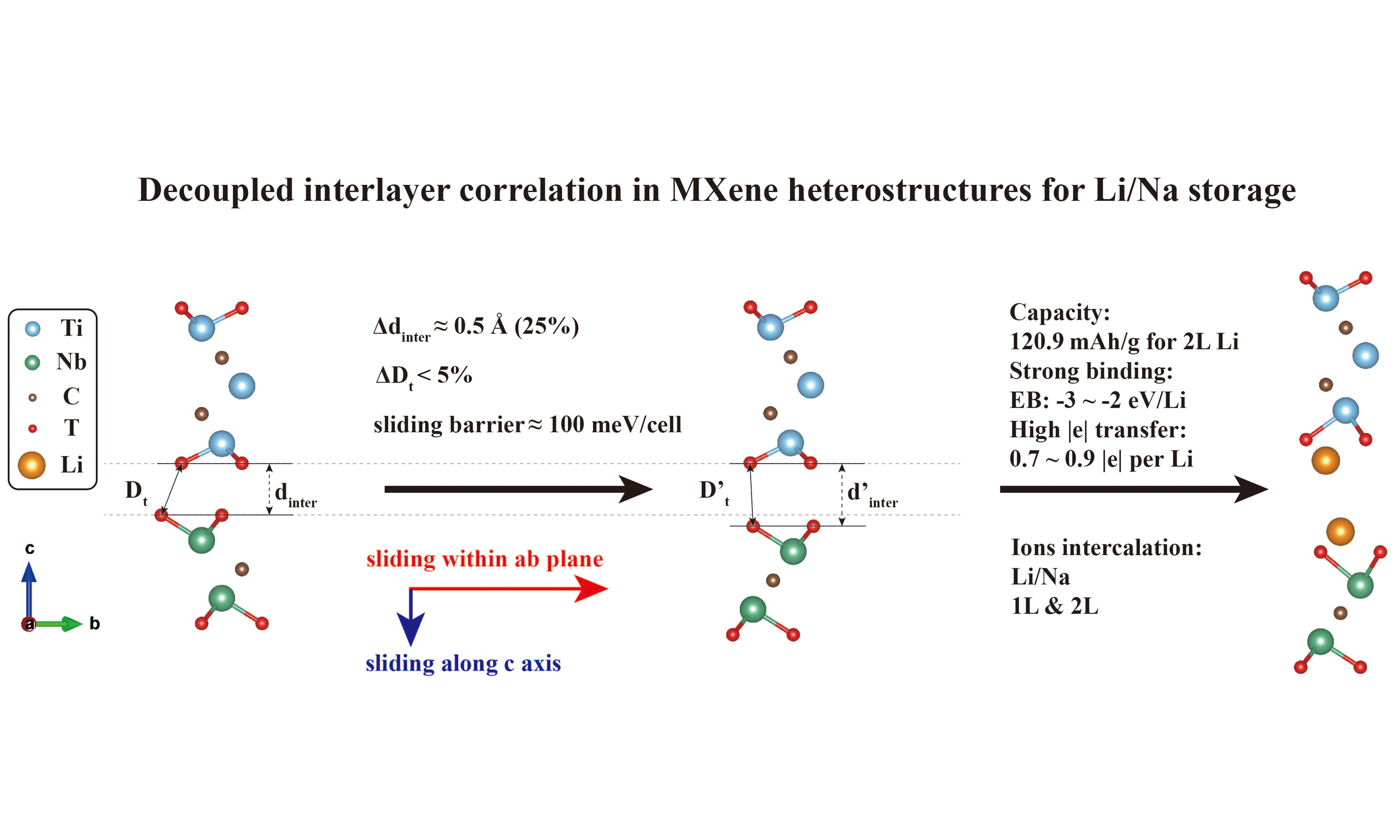
MXene heterostructure compositions offer unique advantages by addressing self-stacking issues and increasing capacity simultaneously. The weak van der Waals (vdW) interaction between MXene heterostructures provides an excellent opportunity for engineering material properties. In this study, using Density Functional Theory (DFT) calculations, we focus on Ti3C2Tx/Nb2CTx heterostructures, discover a synchronous correlation between interlayer spacing and system energy during global sliding, with the distance between the nearest functional groups on opposite sides of the interlayer remaining almost constant, which we term the “decoupled interlayer correlation”. Based on this correlation, we investigate the potential applications of Ti3C2Tx/Nb2CTx 2D heterostructures in batteries and capacitors. Specifically, we perform calculations on 1-layer and 2-layer Li/Na intercalation, focusing on structural transformations and optimal capacities achievable through layer-sliding. Additionally, we explore Young’s moduli of ground-state configurations to characterize the elastic properties. These findings not only provide insights for further research on energy storage utilizing MXene heterostructures but also encourage exploration into sliding in other 2D systems that leverage vdW interactions.
Keywords
INTRODUCTION
The past few decades have witnessed unprecedented population growth and industrial development, leading to rapid depletion of traditional fossil fuels and a host of environmental issues[1-6]. According to the International Energy Agency, global energy demand is projected to rise from 18 TW in 2014 to 24 TW in 2040[7]. Energy security and environmental problems have thus placed humanity in a dilemma. Novel and reliable electrochemical energy-storage technologies are urgently needed, driving the rapid development of devices such as rechargeable batteries and supercapacitors[1,3,5,8].
Lithium-ion batteries (LIBs), first commercialized in the 1990s, have become a mature technology and are now predominantly used in next-generation energy devices[2,6]. Their performance largely depends on the anode materials. Consequently, ultrathin 2D materials, with their atomically thin nanosheets and large surface areas, have been extensively studied for Li storage[1]. Among these, MXenes stand out due to their high aspect ratio, excellent intrinsic electronic and ionic conductivities, good mechanical integrity, easy low-cost “top-down” exfoliation, abundant tunable functional groups, and outstanding hydrophilicity[1,2,4-6,8,9]. These properties make MXenes promising candidates for electrochemical energy storage, as demonstrated by numerous experimental and theoretical studies[10-15].
However, the application of MXenes is often hindered by self-stacking and collapse, which arise from relatively strong van der Waals (vdW) forces and hydrogen bonds between homogeneous layers[16-18]. To address this issue, heterostructures that combine MXenes and other 2D materials, as well as all-MXene multilayer heterostructure films, have been explored because they benefit from weaker interlayer interactions and increased interlayer spacing[19-22]. Since integrating MXenes with other 0D, 1D or 2D materials often involves processing compatibility issues, all-MXene heterostructures were first experimentally created in 2020 using Ti3C2Tx and Nb2CTx[19], and these structures exhibited excellent supercapacitor performance.
However, much remains unknown about the dynamical structural response and underlying mechanisms during alkali atom intercalation and delamination at the atomic scale in MXene heterostructures. Although some experiments have revealed the atomic configuration of several static all-MXene heterostructures[23-25], dynamic atomic-scale characterizations have not yet been achieved, due to the difficulty of preparing perfect all-MXene monolayers and the resolution limits of microscopy[9,26,27]. The bonding conditions, chemical interactions, and the underlying mechanism remains vague. Specifically, the origin of high energy density and high mobility of alkali atoms, the structural characteristics of Li/Na intercalated MXenes, and the structural and electronic interaction between Li/Na and MXenes all require deeper exploration[27,28].
Apart from chemical modification, stack sliding offers a low-cost method to modulate electrical properties by exploiting the weak interlayer vdW interactions in these materials. Such sliding has been shown to achieve reversible polarization switching coupled with lateral motion by overcoming a low energy barrier, leading to the emergence of “slidetronics”[25,29,30]. However, owing to fabrication difficulties, little has been reported, experimentally or theoretically, on interlayer sliding in 2D MXene heterostructures and its related applications[31,32].
Herein, using density functional theory (DFT) calculations, we investigate all-MXene, Ti3C2Tx/Nb2CTx (T denotes terminal functional groups) heterostructures as a model system to determine the most stable configurations for various functional groups and layer-ratios. We then slide one layer relative to the other to examine how the weak vdW interaction modulates the structural and electrical properties. Our results show that interlayer sliding can enlarge the interspace with minimal energy cost, without introducing additional materials. After Li/Na intercalation, this increased spacing effectively reduces volume expansion while barely affecting electron transfer between the intercalated ions and the MXene heterostructures. Thus, the intrinsic weak vdW interaction in Ti3C2Tx/Nb2CTx provides a means to tune the structural properties and, consequently, the electrochemical performance of Li/Na-intercalated MXenes for energy storage. This work is expected to offer valuable insights for the development of MXene-based energy storage technologies using Li/Na ions.
MATERIALS AND METHODS
We performed DFT calculations using the Vienna ab initio simulation package (VASP)[33,34]. The generalized gradient approximation was employed with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional and projector augmented-wave (PAW) potentials[35,36]. A 15 × 15 × 1 Monkhorst-Pack k-meshes and an energy cut-off of 550 eV were used for geometry optimization and the electronic structure calculations. Convergence tests for the cut-off energy and k-points are provided in Supplementary Figure 1. The convergence criteria were set to 0.01 eV/Å for force and 1e-5 eV for energy. Spin polarization was not included, following earlier studies[37-42]. Interlayer vdW interactions were account for using the DFT-D3 method (IVDW = 11)[43].
We considered three terminations (T): -F, -O and -OH, corresponding to Ti3C2F2, Ti3C2O2, Ti3C2(OH)2, Nb2CF2, Nb2CO2 and Nb2C(OH)2. The face-centered cubic (FCC) termination sites, which are the most stable, were chosen for both Ti3C2Tx and Nb2CTx[41,44-47], as illustrated in Figure 1A and B. Our relaxed lattice parameters agree well with previously reported theoretical data [Supplementary Table 1], for example, our calculated a for Ti3C2O2 is 3.024 Å versus reported 3.057 Å[48], confirming the reliability of our computational setup.
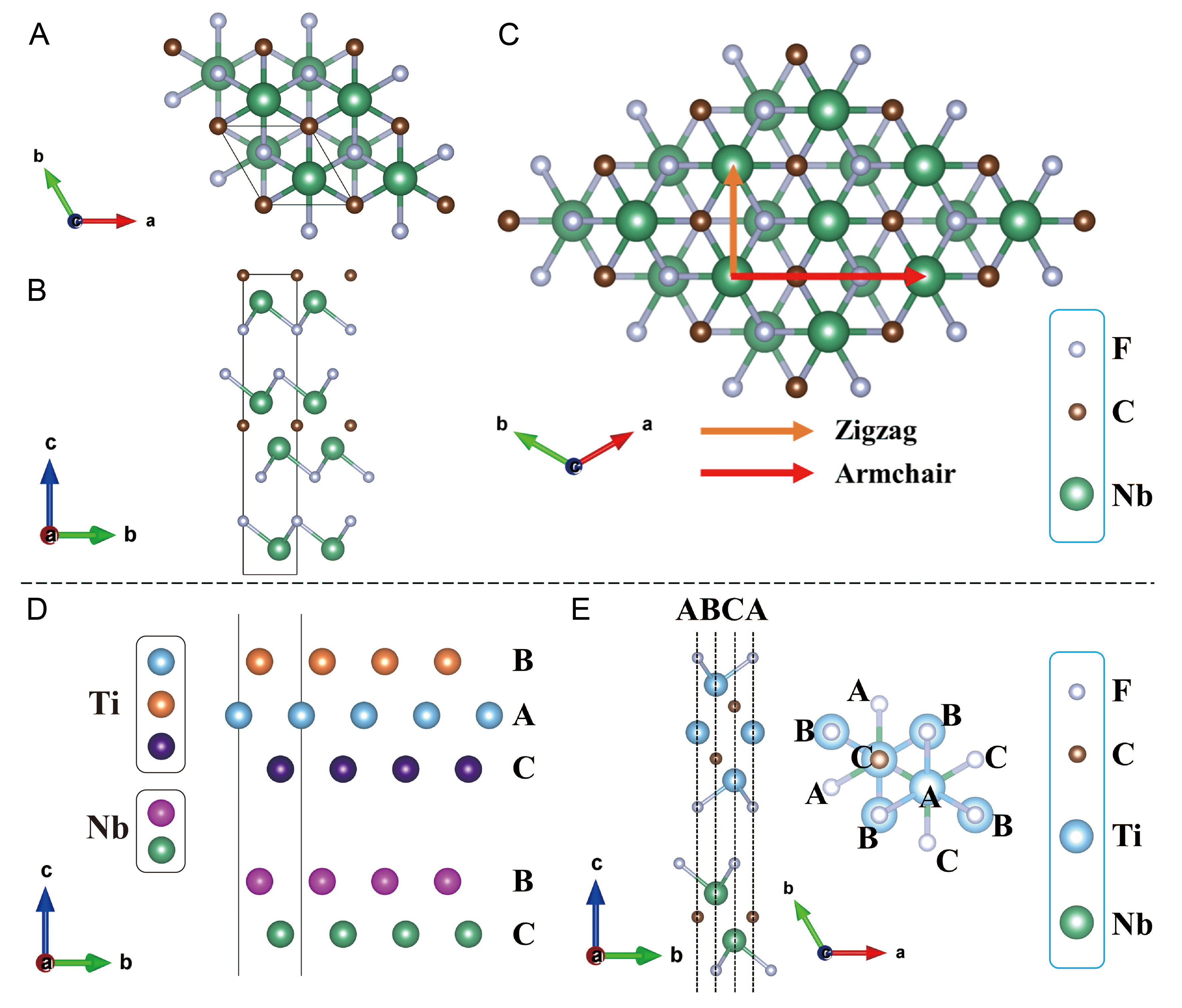
Figure 1. Structural schematics of MXene and MXene heterostructures. (A) Top view of FCC adsorption style in Nb2CF2; (B) Side view of FCC adsorption style in Nb2CTx; (C) Schematic illustration of ZZ (the orange arrow) and AM (the red arrow) sling directions; (D) Initial sliding configuration of Ti3C2Tx/Nb2CTx heterostructure showing metal atoms, the metal atoms exactly locate on three distinctive high-symmetry sites within ab plane denoted as A, B and C; (E) Top and side view of high-symmetry sites A, B and C in initial sliding configuration of Ti3C2F2/Nb2CF2 heterostructure, where every atom locates on one of three high-symmetry sites without exception. FCC: Face-centered cubic; ZZ: zigzag; AM: armchair.
The initial Ti3C2Tx/Nb2CTx heterostructures were constructed by stacking one Ti3C2Tx layer and one Nb2CTx layer (or ratios 1:3 and 3:1). Their in-plane lattice parameters (a, b) were set to the average lengths of the relaxed lattice constants of the individual monolayers, with γ = 120°. This introduces lattice mismatches of approximately 1.35%, 2.30% and 1.61% for -F, -O -OH terminated models, respectively[49]. A vacuum layer of at least 30 Å was added to avoid interactions between neighbouring slabs. A comparison between calculations with and without vdW correction is provided in Supplementary Figure 2.
To assess the effect of lattice mismatch, we estimated the strain energy for each monolayer when constrained to the averaged lattice constant. Specifically, we took the -O terminated 1Ti1Nb heterostructure (which has the largest mismatch) as an example. For each relaxed sliding configuration, we extracted the Ti3C2O2 and Nb2CO2 monolayers and calculated their total energies under both the average lattice constant (E_avg) and the native lattice constant (E_native). The upper limit of strain energy for the heterostructure was then evaluated as ΔE_strain_total = (E_Ti_avg - E_Ti_native) + (E_Nb_avg - E_Nb_native). The calculated strain energies for all 1Ti1Nb -O sliding models are provided in Supplementary Table 2, showing nearly constant during sliding. Additionally, using the ground state configuration AM0ZZδ/6 as an example, we compared the projected density of states (PDOS) of each monolayer part and the T-M (M: metal, i.e., Ti/Nb) part under different lattice conditions [Supplementary Figure 3]. Only minor changes were observed, and the overall metallic character remained intact, which does not affect our conclusions.
In the initial configuration before sliding along the armchair (AM) and zigzag (ZZ) directions [Figure 1C], every atom resides exactly on one of the three high-symmetry sites [Figure 1D and E], where FCC sites correspond to site A for Ti3C2Tx part, and site B or C for Nb2CTx part in Figure 1E. we denote these sites as A, B, and C. Their crystallographic coordinates on the ab plane are
After relaxation, the interlayer spacing (dinter) between Ti3C2Tx and Nb2CTx was defined as the perpendicular distance between two planes that are parallel to the ab plane and each contain the closest atoms of the respective layer. The minimum atomic distance between terminal functional groups from opposite layers (Dt) was also extracted. Definitions of dinter and Dt are illustrated in Supplementary Figure 4. The binding energy of the 1Ti1Nb (1 layer of Ti3C2Tx + 1 layer of Nb2CTx) heterostructure was calculated as EB = EMXene - (ETi3C2Tx + ENb2CTx). Charge transfer between the two layers was analysed using the Bader Charge method[50]. Density of states (DOS) and Band structure data were extracted using vaspkit[51].
For in-plane Young’s moduli calculations, the hexagonal unit cells of MXenes were transformed into rectangular supercells using the transition matrix:
The energy-strain method was adopted. Uniaxial strain along the AM and ZZ directions, as well as biaxial strain, were applied with values of -1.5%, -1%, -0.5%, 0, 0.5%, 1%, 1.5%[52]. After extracting the in-plane elastic constants using vaspkit[51], we calculated the in-plane Young’s modulus (Y2D), shear modulus (G2D), and Poisson’s ratio (υ2D) using following equations[53,54]:
Where x denotes the ZZ direction and y the AM direction.
The volume change (ΔV), thickness change (ΔZ), and area change (ΔS) upon Li/Na intercalation were derived from the atomic positions at the two ends of the heterostructure along the c-axis and from the in-plane lattice vectors (a, b). The binding energy per atom of 1-layer (1L) intercalation was defined as EB = EMXene+1L Li/Na - (ELi/Na + EMXene). For 2-layer (2L) intercalation, the average binding energy per atom was calculated as EB = (EMXene+2L Li/Na - (2 × ELi/Na + EMXene))/2, and the binding energy per atom for the second layer alone was EB = EMXene+2L Li/Na - (ELi/Na + EMXene+1L Li/Na). The alkali ion storage capacities of MXene heterostructures were estimated using the formula[55]:
Where n is the number of intercalated atoms, ZA their valance state, F the Faraday constant (26,801 mAh/mol), MMXene the mole weight of the MXene heterostructure, and M the mole weight of the intercalated metal.
RESULTS AND DISCUSSION
Heterostructures and interlayer sliding
To achieve staggered arrangements of metal atoms, we set BAC-BC stacking Ti3C2Tx/Nb2CTx heterostructures as the initial configurations [Supplementary Figure 5]. We then performed a full-period sliding along both the AM and ZZ directions to search for the most stable structures and to track the variation of system energy during sliding. We considered three terminations (-F, -O and -OH) to thoroughly examine the interfacial structural and electronic properties[56,57]. To further understand the interlayer interaction, three layer-ratios between Ti3C2Tx and Nb2CTx (1:1, 1:3, 3:1) were investigated. In total, we obtained 9 basic configurations, containing 3 surface terminations and 3 layer-ratios, and studied sliding along both the AM and ZZ directions.
Changes of energy and interspace during sliding
Hexagonal 2D crystals exhibit anisotropy in their atomic densities and periodic lengths along the AM and ZZ directions[58-60]. To investigate this phenomenon, Figure 2 shows the evolution of Ti3C2Tx/Nb2CTx heterostructures as they slide along the AM or ZZ directions.
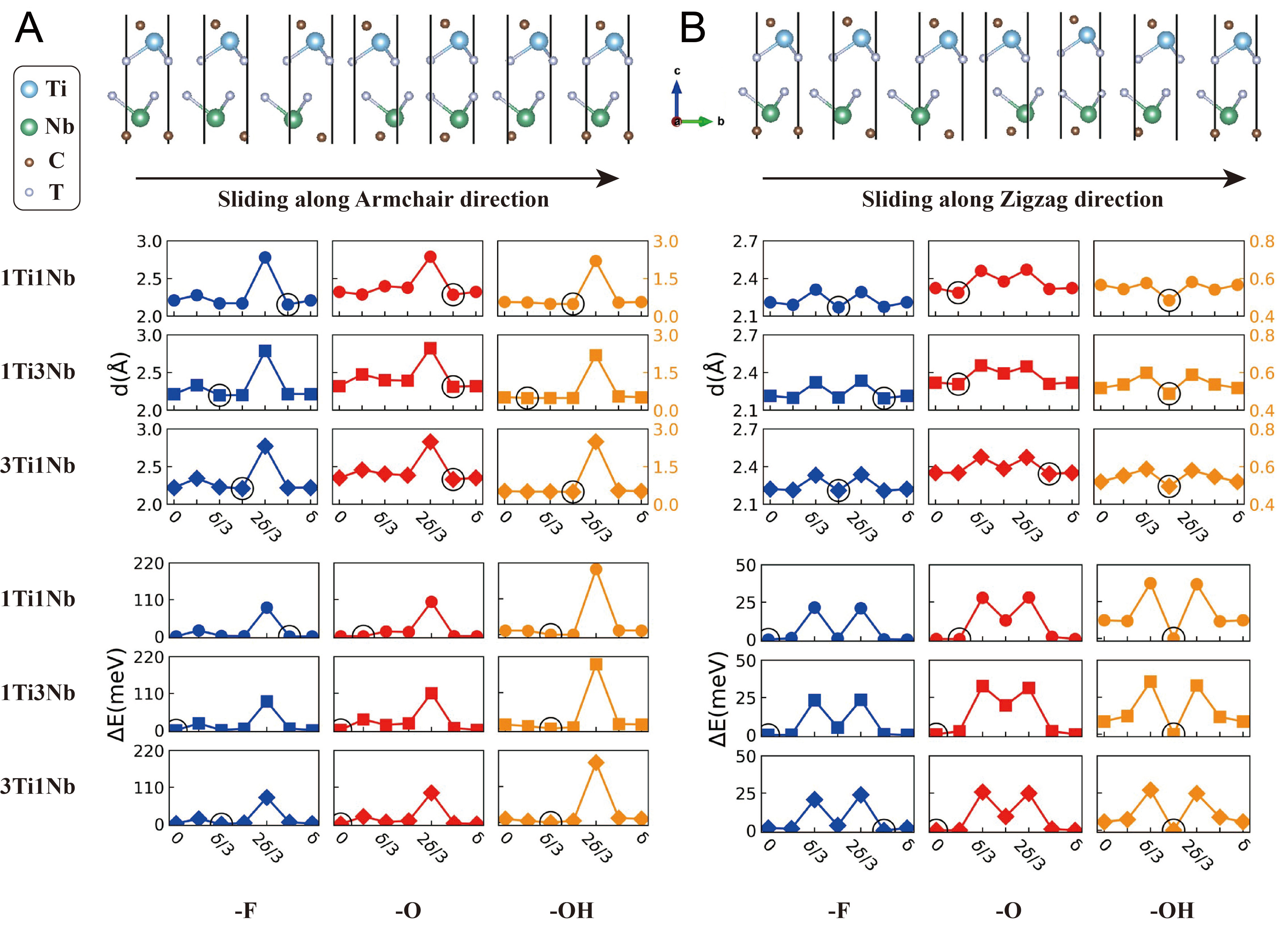
Figure 2. The structural and energetic variations of -F (blue), -O (red) and -OH (orange) terminated Ti3C2Tx/Nb2CTx. The top panel illustrates the atomic configurations during sliding along the AM and ZZ directions. The middle panel shows the interlayer spacing variation for different terminations and layer ratios. The lower panel shows the system energy variation per cell. ΔE is the energy relative to the ground state, and δ is the sliding period. Configurations with minimum interlayer spacings and energies along AM and ZZ are marked by circles. (A) and (B) correspond to sliding along AM and ZZ, respectively. Layer ratios 1:1, 1:3 and 3:1 are denoted by circles, squares and diamonds. The left and right y-axis share the same scale except for -OH terminated models (orange labels). ZZ: Zigzag; AM: armchair.
The upper panels of Figure 2 show the atomic configurations before relaxation. To identify the relative positions of the two layers after relaxation, Supplementary Figure 5 highlights the metal atoms in models with minimum and maximum energies. During sliding, the ground-state configurations (GCs), among the calculated models, are predominantly found at AM0ZZ0 or AMδ/3ZZ0, where the metal atoms (Ti & Nb) adopt an FCC stacking arrangement (BAC-BC or BAC-AB). Conversely, the configurations with maximum-energy configurations (MCs) among the calculated models, occur mainly at AM2δ/3ZZ0, featuring a twin grain boundary (BAC-CA) that forces surface functional groups to face each other directly, leading to a marked increase in system energy.
To determine the preferred relative positions of the two layers in GCs, Supplementary Table 4 summarizes the positions of interlayer Nb atoms relative to Ti3C2Tx. For -O terminated heterostructures, Nb atoms prefer to sit atop the nearest C atoms of Ti3C2Tx, whereas for -OH termination, they prefer atop the surface -OH groups. For -F termination, no clear trend is observed. Detailed structural and energetic data are provided in Appendix I of Supplementary Materials.
The middle panels of Figure 2 show the variation of dinter during sliding. For reference, the dinter values of the pristine Ti3C2Tx and Nb2CTx are as follows: for -F termination, 2.22 and 2.20 Å; for -O, 2.44 and 2.37 Å; for -OH, 0.52 and 0.45 Å, respectively. When sliding occurs along the AM direction, dinter exhibits sharp increases at a displacement of 2δ/3, with increments of approximately 0.55 Å (0.58 Å) for -F, 0.38 Å (0.45 Å) for -O, 1.67 Å (1.75 Å) for -OH relative to the pristine Ti3C2Tx (Nb2CTx). In contrast, along the ZZ direction, dinter shows saddle points at δ/2 between two peaks at δ/3 and 2δ/3, but the increase at the peaks is less than 0.2 Å relative to the pristine Ti3C2Tx (Nb2CTx) for all terminations. Thus, sliding along AM can substantially enlarge dinter compared with pristine MXenes, whereas sliding along ZZ hardly does so.
The lower panels of Figure 2 show the variation in the system energy during sliding. Along AM, the energy peaks at 2δ/3, with energy barriers of about 80 meV/cell (-F), 100 meV/cell (-O), and 200 meV/cell (-OH). Along ZZ, two similar peaks present at δ/3 and at 2δ/3, with energy barriers of 19-23 meV/cell (-F), 23-27 meV/cell (-O), and 27-37 meV/cell (-OH). Thus, sliding along ZZ is energetically preferred over sliding along AM, but it yields a much smaller increase in dinter.
Comparing the middle panel and the lower panel of Figure 2 reveal that dinter and system energy follow similar variation patterns, reaching local extreme at the same sliding positions. To test the generality of this synchronous correlation, we constructed energy contour maps for global sliding [Supplementary Figures 6 and 7], which confirm that GCs always have the smallest dinter and MCs the largest.
To understand the origin of this correlation, we decomposed the system energy into the energies of the Ti3C2Tx, Nb2CTx components and their binding energy EB. Variations in the individual layer energies are negligible comparing with those in EB [Supplementary Tables 5 and 6]. For the case of 1Ti1Nb, Supplementary Figure 8 also presents the striking similarity between EB mappings and global energy mappings. This indicates that the system energy variation is dominated by EB. As shown in Supplementary Figure 9, EB ranges from 15 to 40 meV/Å2 for -F/-O terminated heterostructures, typical of vdW interactions between 2D materials[61,62]. For -OH terminated heterostructures, hydrogen bonding strengthens the interlayer interaction, raising EB to 25-55 meV/Å2.
Here we briefly summarize the effects of layer ratio and termination on sliding behavior. As shown in Supplementary Table 7, varying the layer ratio (1:1, 1:3, 3:1) has little effect on either the sliding energy barrier or the maximum interlayer spacing enlargement (Δd_inter). For example, with -O termination, the barrier changes from 103 meV (1Ti1Nb) to 110 meV (1Ti3Nb) and 93 meV (3Ti1Nb), while Δdinter varies between 0.500 Å and 0.518 Å. Similar insensitivity is observed for -F and -OH terminations. In stark contrast, the termination dictates the sliding behavior: -OH yields barriers about twice as high (~200 meV) and Δdinter three to four times larger (~1.7-2.0 Å) compared with -F/-O (barriers ~80-110 meV, Δdinter ~0.5-0.6 Å). This difference arises because hydrogen bonds in –OH systems introduce strong, directional interlayer interactions that are highly sensitive to sliding displacement, whereas vdW interactions in -F/-O systems are weaker and less stacking-dependent. Thus, layer ratio plays a secondary role; tuning the surface termination is the primary lever for controlling sliding energetics and interlayer spacing in MXene heterostructures.
Decoupled interlayer correlation
The mainly worked vdW interactions in our MXene heterostructure models include direct electrostatic, induction and dispersion interactions[63]. To determine which component dominates the energy variation during sliding, we performed three sets of analyses.
First, we calculated the Bader charges and extracted charge transfer between the two layers [Supplementary Figure 10] and between the nearest atomic groups (Nb&Tx and Ti&Tx) [Supplementary Figure 11]. We then evaluated the correlation between charge transfer and system energy (and dinter) [Supplementary Table 8, Supplementary Figures 12 and 13] to assess the role of direct electrostatic interaction.
Second, we calculated the DOS of GCs and MCs for the whole two layers [Supplementary Figure 14] and for the interlayer atomic groups (Nb&Tx and Ti&Tx) [Supplementary Figure 15], as well as the band structure [Supplementary Figure 16], to evaluate the induction interaction.
Third, we quantified the Pearson coefficients of determination (r) between -1/EB and dinter6, and between -1/EB and Dt6 [Supplementary Tables 6 and 9], to reveal the role of the dispersion interaction.
We now examine each component in turn. First, Bader charge analysis [Supplementary Figures 10 and 11] shows minimal charge transfer between Nb2CTx and Ti3C2Tx during sliding (< 0.06 |e|). Similarly, negligible charge transfer (< 0.1 |e|) is observed between interlayer Ti&Tx and Nb&Tx. Quantitatively, ordinary least squares regression [Supplementary Figures 12 and 13] reveals a poor correlation between charge transfer and system energy (or dinter). These findings contrast with previous studies on MXene heterostructures, where charge transfer played a more important role[48,61]. Therefore, direct electrostatic interaction hardly contributes to the synchronous correlation.
Second, comparison of DOS and band structures between GCs and MCs [Supplementary Figures 14-16] reveals that all heterostructures are metallic. The differences in DOS between GCs and MCs are remarkably subtle [Supplementary Table 10, Supplementary Figures 14 and 15], both for the total DOS of individual MXene layers and for the local DOS (LDOS) of interlayer atoms (Ti&Tx and Nb&Tx), indicating minimal changes in electronic structure. The projected band for interlayer atoms [Supplementary Figure 16] similarly shows negligible difference between GCs and MCs. Furthermore, the distinct peaks in the DOS plots confirm limited electronic interaction between the two layers, consistent with the Bader charge analysis. Thus, induction interaction also barely contributes to the synchronous correlation.
Finally, the Pearson coefficients of determination (r) between -1/EB and dinter6 are all close to 1 (> 0.96 for -F/-O models, > 0.94 for -OH models) with p-value < 1E-25, whereas the r values between -1/EB and Dt6 show no clear trend. We therefore conclude that the interlayer dispersion interaction plays a crucial role in the synchronous trend between system energy and dinter.
To further examine the role of the dispersion interaction, we calculated the distribution of Dt [Supplementary Figures 17-19]. For -F/-O terminations, Dt remains close to its average value across all layer ratios, with a deviation of less than 3.3%. The average Dt values are about 2.8 Å and 2.9 Å, which are roughly twice the vdW radii of F/O[64]. For -OH termination, Dt between interlayer atoms, including -H···-H, -O···-O and -H···-O, varies by less than 10%, except in some models with distorted -OH groups, likely due to intermolecular hydrogen bonding. Notably, for the O atoms in -OH groups, the average Dt (-O···-O) is about 3.0 Å, also close to twice the vdW radii of O[64].
Although Dt varies by less than 5% for -F/-O terminations, dinter can increase by up to 25% relative to its minimum [Supplementary Figure 20]. Specifically, the actual increases in dinter are about 0.6 Å for -F and 0.5 Å for -O terminated heterostructures, respectively. For -OH termination, about 30% increase in Dt can lead to more than 300% increase (about 2.0 Å) in dinter. We term this behavior the “decoupled interlayer correlation”, and illustrate it in Supplementary Figure 21: during sliding, Nb2CTx (or Ti3C2Tx) layer moves not only in-plane but also along c-axis, resulting in a larger dinter while keeping Dt nearly constant. This decoupling between dinter and Dt is the physical origin of the synchronous correlation between dinter and EB. For –OH terminated systems, directional hydrogen bonding alters the interaction mechanism; Dt is no longer constant (variation > 10%), so the “decoupled interlayer correlation” is not strictly applicable.
We hypothesize that the nearly constant Dt minimizes the perturbation of the interfacial electronic states during sliding, as evidenced by the almost unchanged DOS and Bader charge. This “decoupled interlayer correlation”, dominated by isotropic London dispersion interactions, ensures that the local chemical environment and electronic coupling across the interface remain largely unchanged even when the macroscopic layer spacing is manipulated by sliding. Thus, the nearly constant local coordination preserves charge transfer characteristics, explaining why the Bader charge transfer remains stable but correlates poorly with sliding energy or dinter.
To assess the feasibility of achieving such enlarged dinter, we mapped the full-period variation in system energy [Supplementary Figure 22]. The sliding barriers are typically ~100 meV/cell for -O and -F terminated heterostructures, and 200 meV/cell for -OH termination. For context, previously reported sliding barriers for 2D materials range from 0.0045 meV/atom to 15 meV/atom in experiments and theoretical simulations[65-69]. In our study, the global sliding barriers for -O and -F terminated heterostructures range from 3.071 meV/atom to 5.021 meV/atom, which are likely surmountable under practical conditions. For -OH terminated heterostructures, specifically 3Ti1Nb (3 layers of Ti3C2Tx + 1 layer of Nb2CTx), 1Ti3Nb (1 layer of Ti3C2Tx + 3 layers of Nb2CTx) and 1Ti1Nb, the barriers are higher (5.38, 6.57 and 12.545 meV/atom, respectively), making sliding somewhat more challenging.
In summary, the “decoupled interlayer correlation”, arising from interatomic and interlayer vdW interactions, enables the enlargement of dinter while keeping the distances between interlayer functional groups nearly constant. This effect, combined with the metallic conductivity of MXene heterostructures, positions Ti3C2Tx/Nb2CTx heterostructures as a novel class of metallic layered materials with low sliding energy barrier, expanding the family of 2D electronic materials.
Li/Na intercalation
The increase in interlayer spacing achieved at low energy cost can facilitate the intercalation and deintercalation of alkali cations in battery applications. Here, we investigate the potential of Ti3C2Tx/Nb2CTx 2D heterostructures for energy storage by focusing on Li/Na intercalation, due to the low cost and high energy density of these alkali metals[15,70]. Specifically, we calculated binding energies per Li/Na atom and examined structural properties for 1L and 2L Li/Na intercalation, using both GC and MC heterostructures as hosts to explore the effect of enlarged dinter. Three distinct high-symmetry sites in the interlayer were considered: atop terminations, atop C and atop Ti atoms of Ti3C2Tx[71], corresponding to the sites A, B and C respectively. The choice of up to 2L Li/Na intercalation follows established stoichiometry for pure MXenes[55,71,72].
Supplementary Table 11 summarizes the characteristics of the optimized heterostructures. Consistent with experimental observations of Na intercalation in Ti3C2Tx monolayers[72], both the first and second intercalated Li/Na atoms tend to reside directly above the nearest C atoms of either Ti3C2Tx or Nb2CTx. Also, in line with previous studies on pure MXenes, Li/Na adsorption in -F terminated MXenes leads to local geometric distortion that curtails capacity, primarily due to the formation of metal fluorides[71,73].
Figure 3 represents the binding energies and structural changes for 1L Li/Na intercalation in GCs, where Li/Na atoms are located at high-symmetry sites A, B and C as illustrated in Figure 3A. Data are extracted from GCs among the three intercalation sites (A, B and C). Particular attention is paid to the lattice volume change (ΔV), thickness change (ΔZ) and area change (ΔS), relative to heterostructures without intercalation, as these parameters govern the structural response and subsequent battery performance.
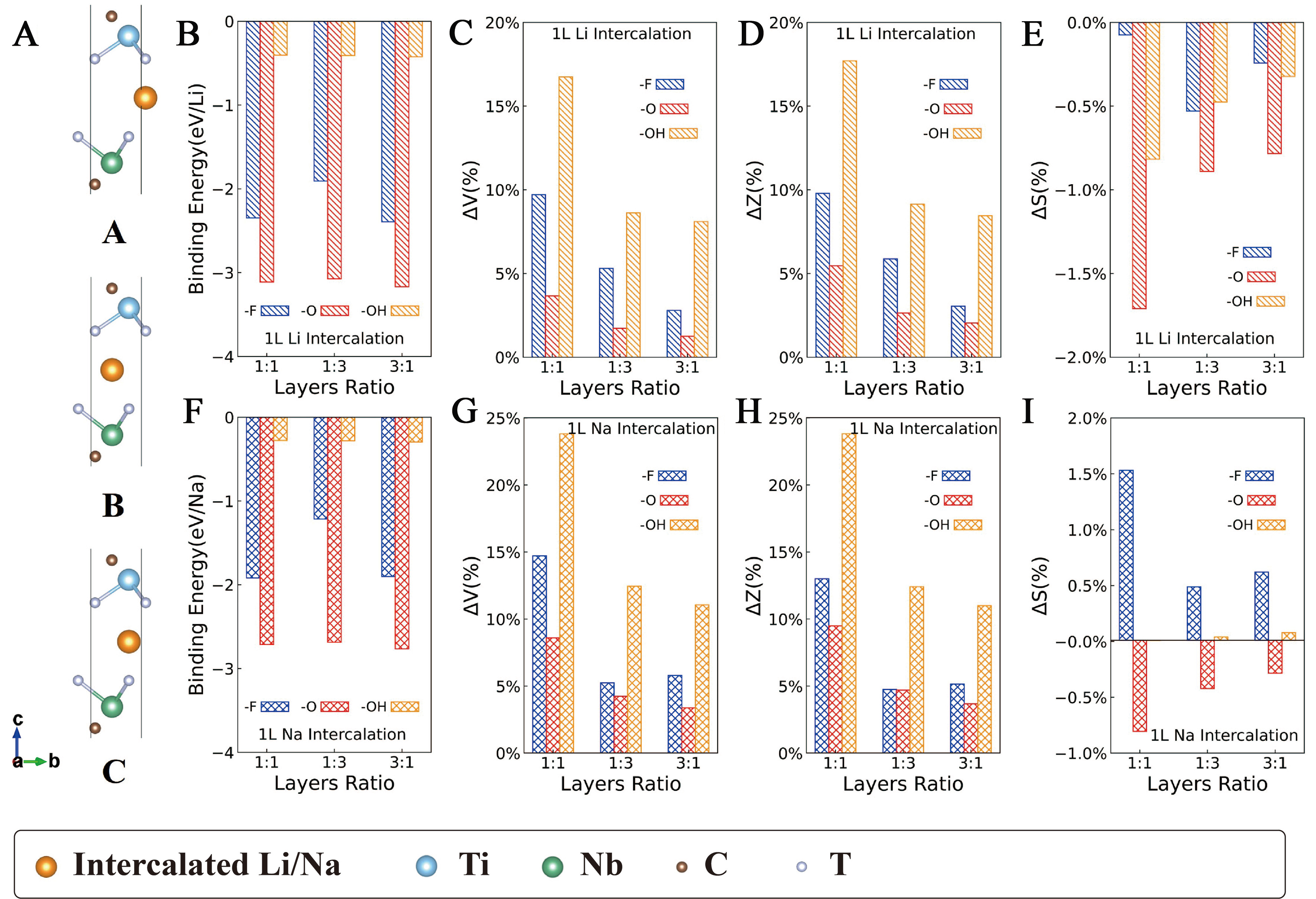
Figure 3. Binding energies and structural changes for 1L Li/Na intercalation in GCs. (A) Schematic diagrams for ground-state MXene heterostructures intercalated by 1L Li at high-symmetry sites A, B and C, respectively, orange balls represent Li/Na atoms. Ground states mostly exhibit in sites B (or A) locating upon C atoms; (B) EB per Li atom for 1L Li intercalation; (C) ΔV, (D) ΔZ and (E) ΔS for 1L Li intercalation; (F) EB per Na atom for 1L Na intercalation; (G) ΔV, (H) ΔZ and (I) ΔS for 1L Na intercalation. (B-E) share the same legend, and so do (F-I). GCs: Ground-state configurations.
-O terminated heterostructures exhibit the strongest binding with Li/Na, followed by -F and then -OH [Figure 3B and F]. For -O termination, EB values are approximately -3.11 eV/Li (1Ti1Nb), -3.07 eV/Li (1Ti3Nb) and -3.17 eV/Li (3Ti1Nb); for Na, they are approximately -2.71, -2.69 and -2.76 eV/Na, respectively. Reported binding (or adsorption) energies for Li in MXene-based layered structures range from 0.146 to -3.573 eV/Li[74-78], and for Na, from 1.36 to -2.74 eV/Na[75,77]. The strong binding observed here thus helps Li/Na clustering, enhancing safety and reversibility. This trend aligns with previous reports that -O functional groups are the most favorable because -O requires two electrons to bond with Li/Na[44]; whereas -F and -OH requires only one; additionally, the H+ in -OH introduces a repulsive force on Li/Na, resulting in less favorable binding energies[71,74,79-81]. For -F terminated heterostructures, EB values are around -2.35 eV/Li (1Ti1Nb), -1.91 eV/Li (1Ti3Nb), -2.39 eV/Li (3Ti1Nb), and the EB of Na are around -1.92 eV/Na (1Ti1Nb), -1.22 eV/Na (1Ti3Nb), -1.90 eV/Na (3Ti1Nb), respectively. While for -OH, the EB values reach up to -0.41 eV/Li and -0.28 eV/Na, indicating much weaker binding. Although -F gives intermediate binding energies, the tendency to form metal fluorides makes -F terminated heterostructures less suitable.
When layer-ratios changes, EB varies little, because Li/Na atoms interact primarily with the interlayer atoms of Ti3C2Tx and Nb2CTx rather than the extra layers in 1Ti3Nb and 3Ti1Nb. This underscores the dominant role of the interface. By the same logic, ΔV, ΔZ and ΔS decrease from 1:1 to 1:3 to 3:1, since Li/Na intercalation only expands the interface region, additional layers add mass without contributing to expansion, so thicker heterostructures expand less.
As shown in Figure 3C-E and 3G-I, structural changes occur predominantly in ΔV and ΔZ, while ΔS remains nearly constant regardless of termination. This indicates that expansion is mainly along the c-axis, reflecting the disparity between strong in-plane ionic bonds and weak out-of-plane vdW forces. For 1L Li intercalation, ΔZ ranges from 2% to 18%, corresponding to a dinter increase of 0.76-2.97 Å; for 1L Na, ΔZ ranges from 9% to 24%, giving a dinter increase of 1.32-3.99 Å. In comparison, experimentally reported dinter increases for Li and Na intercalation in Ti3C2Tx layers are 0.80-2.25 Å and 0.25-2.28 Å, respectively[82]. The larger increment in our work have two origins: first, the heterostructure include a large vacuum, allowing less constrained c-axis expansion than in bulk MXene; second, experimental intercalation may be incomplete (not all interlayers are filled), leading to a smaller measured increment than theoretical prediction.
Results for 1L Li/Na intercalation in MCs are shown in Supplementary Table 12 and Supplementary Figure 23 using same parameters as Figure 3. For -O terminated structures, EB values are approximately -3.21 eV/Li (1Ti1Nb), -3.09 eV/Li (1Ti3Nb) and -3.16 eV/Li (3Ti1Nb); for Na, -2.83, -2.82 and -2.87 eV/Na, respectively. The difference in EB between MCs and GCs is negligible, which we attributed to the “decoupled interlayer correlation”. Regarding structural changes, ΔZ for 1L Li intercalation ranges from 1% to 9% (dinter increase 0.40-0.89 Å), and for Na from 2% to 11% (0.74-1.99 Å); ΔS again remains nearly constant. Notably, ΔV and ΔZ in MCs are generally smaller than those in GCs, and they are halved in some configurations, because the enlarged dinter in MCs provides more space to accommodate intercalated atoms. Thus, the enlarged spacing arising from “decoupled interlayer correlation” has little effect on EB but notably reduces volume expansion during intercalation (from 2%-24% to 1%-12%).
For 2L intercalation in GCs [Figure 4], EB of the 2nd layer Li/Na atoms drops sharply, and ΔZ nearly doubles compared with the 1L case. This sharp decrease in EB arise from enhanced repulsion between the positively charged alkali ions, which substantially limits the capacity[74]. Consequently, intercalation becomes increasingly challenging with higher Li/Na loading. Both ΔV and ΔZ roughly double relative to 1L intercalation. As in the 1L case, -O termination remains the most favorable, and ΔS remain negligible. For 2L Li/Na intercalated in MCs [Supplementary Figure 24], similar trends are observed, but the expansion along c-axis decreases markedly due to enlarged dinter (from 10%-45% in GC cases to 5%-30% in MC cases). Additionally, the EB for Na drops slightly more, likely due to stronger Na+-Na+ repulsion. Detailed structural and energetic data are provided in Appendix II of Supplementary Materials.
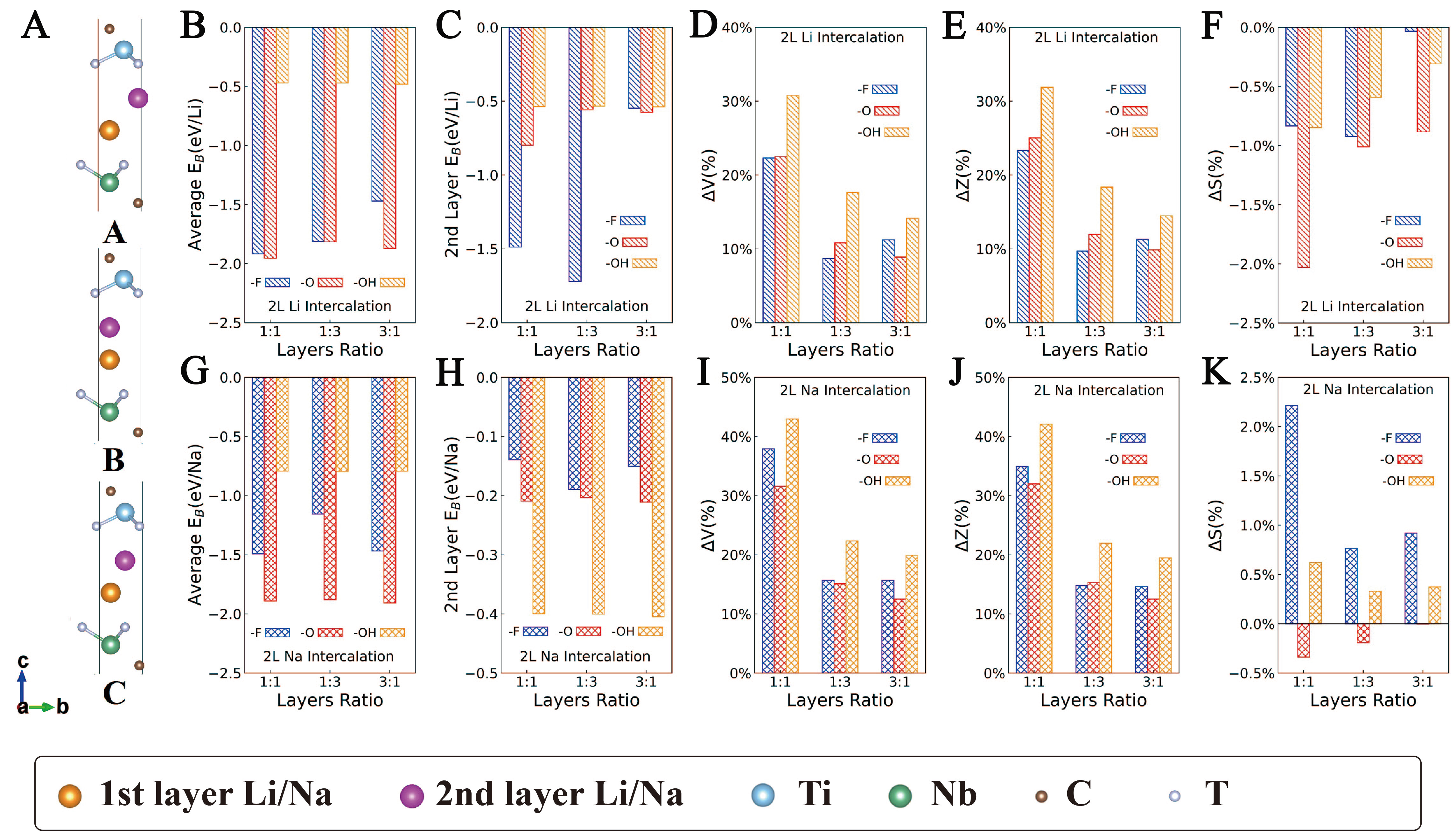
Figure 4. Binding energies and structural changes for 2L Li/Na intercalation in GCs. (A) Schematic diagrams for ground-state MXene heterostructures intercalated by 2L Li/Na, in which the 2nd layer Li/Na atoms locate at high-symmetry sites A, B and C, respectively, orange balls represent the 1st layer Li/Na atoms, and pink balls represent the 2nd layer Li/Na atoms. Ground states mostly exhibit in sites B (or A) locating upon C atoms; (B) EB per Li atom for 2L Li intercalation; (C) EB per Li atom of the 2nd layer intercalated Li; (D) ΔV, (E) ΔZ and (F) ΔS for 2L Li intercalation; (G) EB per Na atom for 2L Na intercalation; (H) EB per Na atom of the 2nd layer intercalated Na, (I) ΔV, (J) ΔZ and (K) ΔS for 2L Na intercalation. (B-F) share the same legend, and so do (G-K). GCs: Ground-state configurations.
Figure 5 shows charge transfer for Li/Na intercalated heterostructures. We focus on the -O terminated 1Ti1Nb heterostructure, which possesses the highest specific capacity (120.90 mAh/g) calculated by equation (4), among the studied layer ratios [Supplementary Table 13], because its 1:1 layer-ratio maximizes the active interface mass fraction, and -O termination gives the strongest binding. For 1L intercalation, similar amounts of electrons are transferred for Li and Na. For 2L case, however, Li donates considerably more charge than Na, as evidenced by isosurface areas shown in Figure 5. Electron transfer occurs predominantly from Li/Na to interlayer O atoms and surface Ti/Nb layers.
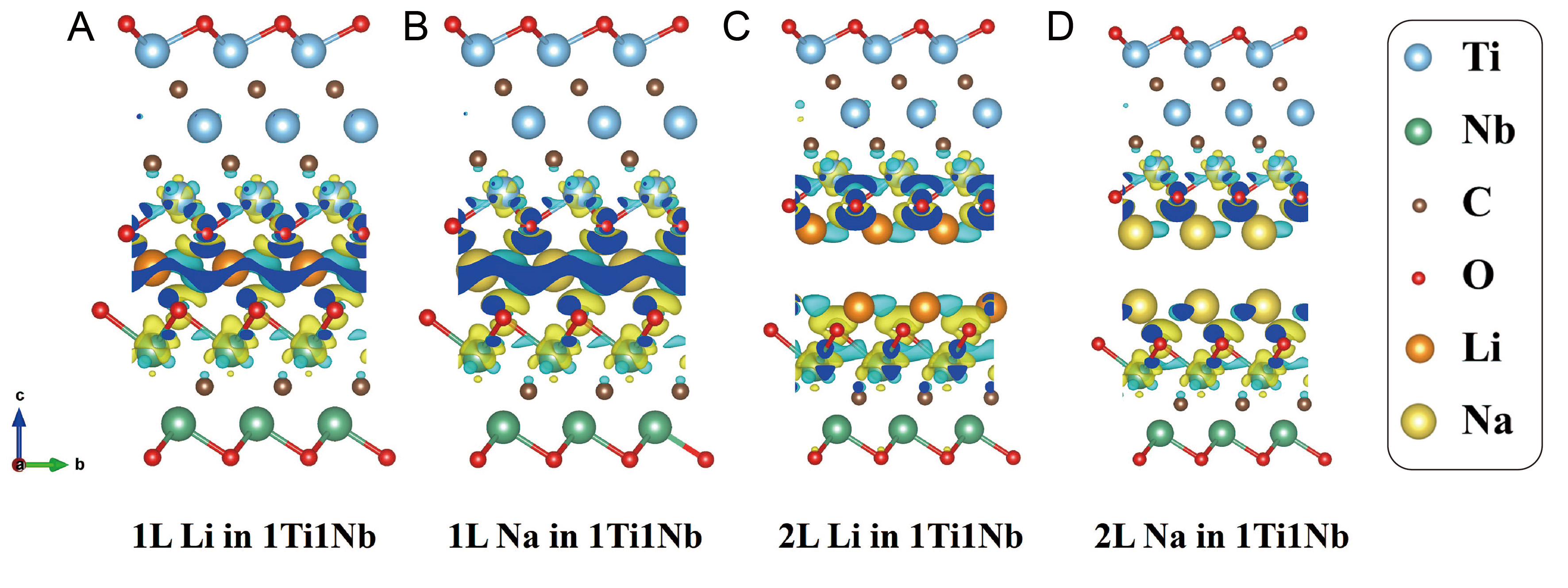
Figure 5. Charge density difference isosurfaces for various alkali intercalation in -O terminated 1Ti1Nb (1 layer of Ti3C2O2 + 1 layer of Nb2CO2), these configurations possess the largest capacities. (A-D) are charge density difference isosurfaces for 1L Li intercalation, 1L Na intercalation, 2L Li intercalation and 2L Na intercalation in 1Ti1Nb, respectively, for 2L intercalation, we regard 2L alkali atoms as a whole. Isosurface value is set at 0.004 e/bohr3: electron accumulation, yellow; electron depletion, cyan.
Bader charge analysis [Supplementary Figure 25] provides quantitative insight. Compared with earlier reports (0.22-0.4 |e| per Li atom)[71], the Li/Na atoms in our heterostructures donate two to three times more electrons: ~0.9 |e| per atom for 1L intercalation (both Li and Na); For 2L intercalation, Li donates 0.7-0.9 |e| per atom, whereas Na donates only 0.1-0.5 |e| per atom. Thus, going from 1L to 2L, Li donates ~50% more electrons, while Na donation remains similar, indicating superior charge transfer capability of Li over Na. These results confirm strong Coulombic interaction between the alkali atoms and heterostructures, effectively preventing agglomeration.
Supplementary Figures 26-29 summarize the electron transfer from Li/Na atoms to the Ti3C2Tx and Nb2CTx components in both GCs and MCs for various terminations and layer-ratios. We highlight three key factors:
(1) alkali identity: Li donates more electrons than Na, especially for -OH termination. Na donates electrons more evenly to the two MXene layers. Li also tends to form fluorides with -F, leading to imbalanced electron transfer.
(2) Number of layers: From 1L to 2L, Li donates 50% more electrons, whereas Na donation remains nearly unchanged, indicating limited electron-donating capability of Na at higher loading.
(3) Configuration (GC vs. MC): The total electron donation is similar for GCs and MCs, showing that enlarged dinter has minimal effect on Coulombic interaction.
The energies for configurations with different intercalation sites (A, B, C) differ negligibly (typically < 1 eV/cell) for both Li and Na and for both 1L and 2L intercalation [Supplementary Figure 30]. This suggests that the intercalated Li/Na atoms can easily move among high-symmetry sites, implying high mobility. This is consistent with previous reports of low diffusion barriers for Li/Na on MXene monolayers[71,72,81].
Layer ratio barely affects charge transfer but plays a vital role in capacity. The specific capacity (gravimetric) decreases monotonically with increasing number of inactive layers: 1Ti1Nb -O has the highest value (120.9 mAh/g for 2L Li), followed by 3Ti1Nb (~63.6 mAh/g) and 1Ti3Nb (~59.4 mAh/g) with larger thickness, consistent with reported trends in Supplementary Table 13. Our capacities are lower than those of typical pure MXenes (e.g., 250 mAh/g for Ti3C2O2)[55,74] because only the Ti3C2Tx/Nb2CTx interface stores Li/Na; additional layers add mass without providing new storage sites. Accordingly, the volume expansion (ΔZ) upon intercalation follows the same order: 1Ti1Nb expands the most (up to ~18% for 1L Li), while 3Ti1Nb expands the least (up to ~8%). The binding energy (EB) remains nearly constant across layer ratios (variation < 0.2 eV/atom for -O/-OH models), confirming that the interlayer chemical environment is dominated by the immediate interface.
Young’s modulus
In addition to electrochemical performance, the application of flexible devices also requires the evaluation of their mechanical properties. Based on standard energy-strain method as detailed in prior MXene studies[52,83], we evaluate the in-plane stiffness (Y2D, in N/m) of the ground-state Ti3C2Tx/Nb2CTx heterostructures [Supplementary Tables 14 and 15]. To avoid the ambiguity of defining a “thickness” for heterostructures with a vacuum layer, we focus on Y2D rather than the bulk-equivalent Young’s modulus E (GPa) in our discussion, following the convention recommended for 2D materials[83].
The calculated Y2D values for -O terminated heterostructures are higher than those for -F and -OH. For instance, Y2D of the robust -O terminated 1Ti1Nb heterostructure reaches 583 N/m (along y-direction), while its -F and -OH counterparts give 445 N/m and 467 N/m, respectively. This trend, with O-termination yielding the highest stiffness, agrees well with previous findings on pristine MXenes[52,54,83,84]. This difference could be attributed to the robust O–M bonds, as –O exhibits higher coordination and obtains more electron (~1.1 |e|) with metal atoms due to its higher coordination compared to -F (~0.75 |e|) and -OH (~0.74 |e|) [Supplementary Table 16], as evidenced by the larger M–O bond stiffness[47,83]. Notably, Y2D for -F and -OH terminated structures are consistently similar across all stacking configurations, matching the reported statistical equivalence of F- and OH-termination strengthening effects in pure MXenes[83].
Furthermore, the Y2D values of 1Ti1Nb heterostructures (436-584 N/m) are obviously lower than those of high-stiffness pure MXenes like Nb4C3O2 (605.99 N/m)[52]. This is expected because the heterostructures are effectively multilayers with weak vdW interlayer coupling, leading to a higher bending rigidity and structural thickness, while their in-plane load-bearing capacity (Y2D) is still dominated by the sum of individual stiff layers. The observed trend confirms that the in-plane stiffness of MXenes is a strong function of the overall layer count and surface termination composition, as determined by the number and strength of the M–T bonds within the framework[52,83], the flexibility diminishes as the atomic layer thickness of MXene increases, aligning with earlier studies[52,84].
The calculated Y2D values [Supplementary Table 14] increase systematically with total layer count: 3Ti1Nb (four layers total) shows the highest stiffness (e.g., 1,260.65 N/m for -O), followed by 1Ti3Nb (1,142.99 N/m for -O) and 1Ti1Nb (583.69 N/m for -O). This trend is expected because Y2D (in N/m) is a width-normalized force constant; adding more layers of stiff MXene in parallel increases the force required to achieve a given strain. When normalized by effective thickness (i.e., equivalent 3D modulus), the multilayers become softer due to weak interlayer coupling - a point already discussed above.
Discussion
The synchronous correlation between interlayer spacing (dinter) and binding energy (EB) during sliding arises from a “decoupled interlayer correlation”: the distance between nearest functional groups (Dt) remains nearly constant, while dinter varies by up to 25% for -F/-O termination. This decoupling, governed by isotropic London dispersion forces, allows remarkable tuning of interlayer spacing (~0.5 Å for -F/-O, ~1.7-2.0 Å for -OH) with minimal perturbation to electronic structure and low sliding barriers (3-12 meV/atom). Therefore, for applications requiring large interlayer expansion (e.g., ion intercalation), -OH termination is advantageous despite its higher barrier; for low-energy sliding manipulation, -F or -O termination is preferred. The weak dependence on layer ratio simplifies device fabrication because the interface chemistry dominates.
Practical MXene surfaces inevitably contain defects and mixed terminations, and operating temperatures vary[85]. Encouragingly, a recent high-throughput study[86] on 230 2D materials confirmed a universal negative correlation between interlayer binding energy and equilibrium distance, which was further validated by room-temperature atomic force microscope (AFM) on C, BN, and In2Se3. These findings support that the synchronous correlation we discovered is robust against moderate temperature variations and realistic surface imperfections, reinforcing its relevance for real-world devices.
Compared with sliding in some MXene homo-structures (barriers ~80-130 meV/atom) and MXene/MoS2 heterostructures (barriers ~15-20 meV/atom)[69], the barriers in Ti3C2Tx/Nb2CTx (3-12 meV/atom) are substantially lower, while the unique decoupling between dinter and Dt has not been reported in those systems. Unlike homogeneous MXene sliding, where functional groups face identical chemical environments, the heterostructure introduces asymmetric interfaces that stabilize the constant Dt across a wide sliding range. This work thus expands the “slidetronics” paradigm to all-MXene heterostructures.
Turning to energy storage applications, for Li/Na storage, Ti3C2O2/Nb2CO2 offers strong Coulombic interaction (0.7-0.9 |e| per Li) and reduced volume expansion in MCs, but the specific capacity (~120 mAh/g for 2L Li) is moderate due to the heterostructure’s large molar mass. A 1:1 layer ratio maximizes capacity, while asymmetric ratios (e.g., 3Ti1Nb) provide higher in-plane stiffness (~1,261 N/m) for structural reinforcement.
Before concluding, we acknowledge several simplifications in our study. The assumption of ideal FCC termination ordering is a pragmatic starting point, yet real MXene surfaces typically exhibit a mixture of -F, -O, and -OH groups. Static calculations of Li/Na intercalation provide binding energies but do not capture finite-temperature diffusion dynamics; future work should therefore employ kinetic Monte Carlo or molecular dynamics to assess rate performance. Similarly, the 0 K sliding barriers reported here may be overestimates, as thermal activation could facilitate interlayer motion under operating conditions. On the experimental side, in situ X-Ray diffraction (XRD) or transmission electron microscope (TEM) characterization during sliding would be invaluable for verifying the predicted decoupling. More broadly, we suggest that the “decoupled interlayer correlation” is not unique to MXenes and may be exploitable in other vdW heterostructures, such as transition metal dichalcogenides, for slidetronics or energy storage.
CONCLUSIONS
This work systematically investigated, via DFT calculations, the interlayer sliding behavior and its role in Li/Na ion storage for Ti3C2Tx/Nb2CTx MXene heterostructures. We uncovered a synchronous correlation between interlayer spacing and system energy during sliding, which originates from the coupling of interlayer and interatomic vdW interactions - termed the “decoupled interlayer correlation” - enabling substantial tuning of interlayer spacing with minimal perturbation to the electronic structure. The enlarged spacing thus achieved effectively mitigates the volume expansion upon alkali-ion intercalation while preserving strong binding and high charge transfer, highlighting a promising strategy for flexible energy-storage interfaces. Looking forward, the concept of sliding-induced property modulation demonstrated here is not limited to MXenes; recent advances have shown that similar sliding-strain synergy can break symmetry and realize emergent states such as non-alter spin splitting in other vdW systems[87], suggesting that the “decoupled interlayer correlation” and related mechanisms could be extended to multifunctional devices including spintronics and multiferroic memories.
DECLARATIONS
Acknowledgments
This research work was supported by the Big Data Computing Center of Southeast University for providing high-performance computing clusters and technical support, and the Center for Fundamental and Interdisciplinary Sciences of Southeast University for administrative and facility support.
Authors’ contributions
Made substantial contributions to conception and design of the study: Zhu, C.; Sun, W.; Yuan, D.
Performed data analysis and interpretation: Yuan, D.
Drafting the work: Yuan, D.
Revising the work: Zhu, C.; Sun, W.
Supervision: Zhu, C.; Sun, L.; Sun, W.
Technical support: Xiong, Y.
Availability of data and materials
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author(s).
AI and AI-assisted tool statement
During the preparation of this manuscript, the AI tool Deepseek (version 3.2, released 2025-12-01) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
The authors acknowledge the financial support provided by the National Natural Science Foundation of China (Nos. 12274067 and 92464101), the open research fund of Suzhou Laboratory (No. SZLAB-1608-2024-TS019).
Conflicts of interest
Sun, L. is a Senior Editorial Board Member of the journal Microstructures. Zhu, C. is a Guest Editor of the Special Issue “Functional Microstructures in Advanced Porous and 2D Materials” of the journal Microstructures. Sun, L. and Zhu, C. were not involved in any steps of editorial processing, notably including reviewers’ selection, manuscript handling and decision making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Rahman, M. M.; Imani, S.; Anjum, N.; Sijuade, A. A.; Okoli, O. Materials and design strategies for next-generation energy storage: a review. Renew. Sustain. Energy. Rev. 2025, 212, 115368.
2. Zheng, C.; Yao, Y.; Rui, X.; et al. Functional MXene‐based materials for next‐generation rechargeable batteries. Adv. Mater. 2022, 34, 2204988.
3. Xi, W.; Jin, J.; Zhang, Y.; et al. Hierarchical MXene/transition metal oxide heterostructures for rechargeable batteries, capacitors, and capacitive deionization. Nanoscale 2022, 14, 11923-44.
4. Gao, M.; Wang, F.; Yang, S.; et al. Engineered 2D MXene-based materials for advanced supercapacitors and micro-supercapacitors. Mater. Today. 2024, 72, 318-58.
5. Rashid Khan, H.; Latif Ahmad, A. Supercapacitors: overcoming current limitations and charting the course for next-generation energy storage. J. Ind. Eng. Chem. 2025, 141, 46-66.
6. An, Y.; Tian, Y.; Shen, H.; Man, Q.; Xiong, S.; Feng, J. Two-dimensional MXenes for flexible energy storage devices. Energy. Environ. Sci. 2023, 16, 4191-250.
7. Shinde, P. A.; Patil, A. M.; Lee, S.; Jung, E.; Chan Jun, S. Two-dimensional MXenes for electrochemical energy storage applications. J. Mater. Chem. A. 2022, 10, 1105-49.
8. Liu, W.; Cao, J.; Song, F.; et al. A double transition metal Ti2NbC2Tx MXene for enhanced lithium-ion storage. Rare. Met. 2023, 42, 100-10.
9. Sobyra, T. B.; Matthews, K.; Mathis, T. S.; Gogotsi, Y.; Fenter, P. Operando X-ray reflectivity reveals the dynamical response of Ti3C2 MXene film structure during electrochemical cycling. ACS. Energy. Lett. 2022, 7, 3612-7.
10. Guo, Y.; Liu, D.; Huang, B.; Wang, L.; Xia, Q.; Zhou, A. Effects of surface compositions and interlayer distance on electrochemical performance of Mo2CTX MXene as anode of Li-ion batteries. J. Phys. Chem. Solids. 2023, 176, 111238.
11. Zhou, Y.; Yin, L.; Xiang, S.; et al. Unleashing the potential of MXene‐based flexible materials for high‐performance energy storage devices. Adv. Sci. 2024, 11, e2304874.
12. Gokul Eswaran, S.; Rashad, M.; Santhana Krishna Kumar, A.; El‐Mahdy, A. F. M. A comprehensive review of Mxene‐based emerging materials for energy storage applications and future perspectives. Chem. Asian. J. 2025, 20, e202401181.
13. Tang, X.; Guo, X.; Wu, W.; Wang, G. 2D metal carbides and nitrides (MXenes) as high‐performance electrode materials for lithium‐based batteries. Adv. Energy. Mater. 2018, 8, 1801897.
14. Ma, P.; Fang, D.; Liu, Y.; Shang, Y.; Shi, Y.; Yang, H. Y. MXene‐based materials for electrochemical sodium‐ion storage. Adv. Sci. 2021, 8, e2003185.
15. Aslam, M. K.; Niu, Y.; Xu, M. MXenes for Non‐Lithium‐Ion (Na, K, Ca, Mg, and Al) Batteries and Supercapacitors. Adv. Energy. Mater. 2020, 11, 2000681.
16. Li, X.; Ran, F.; Yang, F.; Long, J.; Shao, L. Advances in MXene films: synthesis, assembly, and applications. Trans. Tianjin. Univ. 2021, 27, 217-47.
17. Shen, X.; Xiong, Y.; Hai, R.; Yu, F.; Ma, J. All-MXene-Based integrated membrane electrode constructed using Ti3C2TX as an intercalating agent for high-performance desalination. Environ. Sci. Technol. 2020, 54, 4554-63.
18. Tang, J.; Huang, X.; Qiu, T.; et al. Interlayer space engineering of MXenes for electrochemical energy storage applications. Chem. Eur. J. 2021, 27, 1921-40.
19. Li, Z.; Dall’agnese, Y.; Guo, J.; Huang, H.; Liang, X.; Xu, S. Flexible freestanding all-MXene hybrid films with enhanced capacitive performance for powering a flex sensor. J. Mater. Chem. A. 2020, 8, 16649-60.
20. Bandaru, N.; Reddy, C. V.; Vallabhudasu, K.; et al. Exploring the potential of MXene nanohybrids as high-performance anode materials for lithium-ion batteries. Chem. Eng. J. 2024, 500, 157317.
21. Hussain, I.; Lamiel, C.; Javed, M. S.; et al. MXene-based heterostructures: Current trend and development in electrochemical energy storage devices. Progr. Energy. Combust. Sci. 2023, 97, 101097.
22. Ding, T.; Jiang, X.; Quan, J.; et al. Recent progress in two-dimensional van der Waals heterojunctions for flexible energy storage applications. Adv. Compos. Hybrid. Mater. 2025, 8, 324.
23. Kamysbayev, V.; Filatov, A. S.; Hu, H.; et al. Covalent surface modifications and superconductivity of two-dimensional metal carbide MXenes. Science 2020, 369, 979-83.
24. Li, M.; Li, X.; Qin, G.; et al. Halogenated Ti3C2 MXenes with electrochemically active terminals for high-performance zinc ion batteries. ACS. Nano. 2021, 15, 1077-85.
25. Nasrin, K.; Sudharshan, V.; Arunkumar, M.; Sathish, M. 2D/2D Nanoarchitectured Nb2C/Ti3C2 MXene heterointerface for high-energy supercapacitors with sustainable life cycle. ACS. Appl. Mater. Interfaces. 2022, 14, 21038-49.
26. de Kogel, A.; Wang, R. J.; Tsai, W. Y.; et al. Material characterization methods for investigating charge storage processes in 2D and layered materials-based batteries and supercapacitors. Nanoscale 2025, 17, 13531-60.
27. Li, G.; Boulanger, N.; Gurzęda, B.; Bi, S.; Hennig, C.; Talyzin, A. V. Operando X‐Ray diffraction study of MXene electrode structure in supercapacitors with alkali metal electrolytes. Small. Science. 2025, 5, e202500367.
28. Li, X.; Huang, Z.; Shuck, C. E.; Liang, G.; Gogotsi, Y.; Zhi, C. MXene chemistry, electrochemistry and energy storage applications. Nat. Rev. Chem. 2022, 6, 389-404.
29. Vizner Stern, M.; Waschitz, Y.; Cao, W.; et al. Interfacial ferroelectricity by van der Waals sliding. Science 2021, 372, 1462-6.
30. Yasuda, K.; Wang, X.; Watanabe, K.; Taniguchi, T.; Jarillo-Herrero, P. Stacking-engineered ferroelectricity in bilayer boron nitride. Science 2021, 372, 1458-62.
31. Arole, K.; Pas, S. E.; Thakur, R. M.; et al. Effects of intercalation on ML-Ti3C2Tz MXene properties and friction performance. ACS. Appl. Mater. Interfaces. 2024, 16, 64156-65.
32. Feng, Q.; Dou, M.; Yang, J.; et al. Heterostructures comprised of MXene nanosheets for tribology: a review. ACS. Appl. Nano. Mater. 2024, 7, 22379-416.
33. Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996, 54, 11169-86.
34. Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15-50.
35. Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865-8.
37. Gao, G.; O’Mullane, A. P.; Du, A. 2D MXenes: a new family of promising catalysts for the hydrogen evolution reaction. ACS. Catal. 2016, 7, 494-500.
38. Khazaei, M.; Ranjbar, A.; Arai, M.; Sasaki, T.; Yunoki, S. Electronic properties and applications of MXenes: a theoretical review. J. Mater. Chem. C. 2017, 5, 2488-503.
39. Hu, J.; Xu, B.; Ouyang, C.; Zhang, Y.; Yang, S. A. Investigations on Nb2C monolayer as promising anode material for Li or non-Li ion batteries from first-principles calculations. RSC. Adv. 2016, 6, 27467-74.
40. Zhao, S.; Kang, W.; Xue, J. Manipulation of electronic and magnetic properties of M2C (M = Hf, Nb, Sc, Ta, Ti, V, Zr) monolayer by applying mechanical strains. Appl. Phys. Lett. 2014, 104, 133106.
41. Khazaei, M.; Arai, M.; Sasaki, T.; et al. Novel electronic and magnetic properties of two‐dimensional transition metal carbides and nitrides. Adv. Funct. Mater. 2012, 23, 2185-92.
42. Gandi, A. N.; Alshareef, H. N.; Schwingenschlögl, U. Thermoelectric performance of the MXenes M2CO2 (M = Ti, Zr, or Hf). Chem. Mater. 2016, 28, 1647-52.
43. Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456-65.
44. Hu, T.; Hu, M.; Gao, B.; Li, W.; Wang, X. Screening surface structure of MXenes by high-throughput computation and vibrational spectroscopic confirmation. J. Phys. Chem. C. 2018, 122, 18501-9.
45. Seh, Z. W.; Fredrickson, K. D.; Anasori, B.; et al. Two-dimensional molybdenum carbide (MXene) as an efficient electrocatalyst for hydrogen evolution. ACS. Energy. Lett. 2016, 1, 589-94.
46. Zhan, C.; Sun, W.; Kent, P. R. C.; Naguib, M.; Gogotsi, Y.; Jiang, D. Computational screening of MXene electrodes for pseudocapacitive energy storage. J. Phys. Chem. C. 2018, 123, 315-21.
47. Luo, K.; Zha, X. H.; Zhou, Y.; Huang, Q.; Zhou, S.; Du, S. Theoretical exploration on the vibrational and mechanical properties of M3C2/M3C2T2 MXenes. Int. J. Quantum. Chem. 2020, 120, e26409.
48. Xu, L.; Wu, T.; Kent, P. R. C.; Jiang, D. Interfacial charge transfer and interaction in the MXene/TiO2 heterostructures. Phys. Rev. Materials. 2021, 5, 054007.
49. Christensen, A.; Carter, E. A. Adhesion of ultrathin ZrO2(111) films on Ni(111) from first principles. J. Chem. Phys. 2001, 114, 5816-31.
50. Yu, M.; Trinkle, D. R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 2011, 134, 064111.
51. Wang, V.; Xu, N.; Liu, J. C.; Tang, G.; Geng, W. T. VASPKIT: a user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033.
52. Hu, T.; Yang, J.; Li, W.; Wang, X.; Li, C. M. Quantifying the rigidity of 2D carbides (MXenes). Phys. Chem. Chem. Phys. 2020, 22, 2115-21.
53. Imani Yengejeh, S.; Kazemi, S. A.; Wen, W.; Wang, Y. Multiscale numerical simulation of in-plane mechanical properties of two-dimensional monolayers. RSC. Adv. 2021, 11, 20232-47.
54. Fu, Z. H.; Zhang, Q. F.; Legut, D.; et al. Stabilization and strengthening effects of functional groups in two-dimensional titanium carbide. Phys. Rev. B. 2016, 94, 104103.
55. Xie, Y.; Dall’Agnese, Y.; Naguib, M.; et al. Prediction and characterization of MXene nanosheet anodes for non-lithium-ion batteries. ACS. Nano. 2014, 8, 9606-15.
56. Anasori, B.; Lukatskaya, M. R.; Gogotsi, Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2017, 2, 16098.
57. Pomerantseva, E.; Gogotsi, Y. Two-dimensional heterostructures for energy storage. Nat. Energy. 2017, 2, 17089.
58. Mortazavi, B.; Rabczuk, T. Anisotropic mechanical properties and strain tuneable band-gap in single-layer SiP, SiAs, GeP and GeAs. Physica. E. 2018, 103, 273-8.
59. Arab, A.; Li, Q. Anisotropic thermoelectric behavior in armchair and zigzag mono- and fewlayer MoS2 in thermoelectric generator applications. Sci. Rep. 2015, 5, 13706.
60. Naderi, S.; Javaheri, S.; Shahrokhi, M.; Nia, B. A.; Shahmoradi, S. Optical properties of zigzag and armchair ZnO nanoribbons. Physica. E. 2020, 124, 114218.
61. Li, R.; Sun, W.; Zhan, C.; Kent, P. R. C.; Jiang, D. Interfacial and electronic properties of heterostructures of MXene and graphene. Phys. Rev. B. 2019, 99, 085429.
62. Björkman, T.; Gulans, A.; Krasheninnikov, A. V.; Nieminen, R. M. van der Waals bonding in layered compounds from advanced density-functional first-principles calculations. Phys. Rev. Lett. 2012, 108, 235502.
63. Kaplan, I. G. Types of Intermolecular Interactions: Qualitative Picture. In Intermolecular Interactions; 2006; pp 25-79.
65. Li, L.; Wu, M. Binary compound bilayer and multilayer with vertical polarizations: two-dimensional ferroelectrics, multiferroics, and nanogenerators. ACS. Nano. 2017, 11, 6382-8.
66. Yang, Q.; Wu, M.; Li, J. Origin of two-dimensional vertical ferroelectricity in WTe2 bilayer and multilayer. J. Phys. Chem. Lett. 2018, 9, 7160-4.
67. Lu, P.; Kim, J. S.; Yang, J.; et al. Origin of superconductivity in the Weyl semimetal WTe2 under pressure. Phys. Rev. B. 2016, 94, 224512.
68. Hu, H.; Sun, Y.; Chai, M.; Xie, D.; Ma, J.; Zhu, H. Room-temperature out-of-plane and in-plane ferroelectricity of two-dimensional β-InSe nanoflakes. Appl. Phys. Lett. 2019, 114, 252903.
69. Zhang, Y.; Chen, X. Arramel; et al. Atomic-scale superlubricity in Ti2CO2@MoS2 layered heterojunctions interface: a first principles calculation study. ACS. Omega. 2021, 6, 9013-9.
70. Das, P.; Wu, Z. S. MXene for energy storage: present status and future perspectives. J. Phys. Energy. 2020, 2, 032004.
71. Tang, Q.; Zhou, Z.; Shen, P. Are MXenes promising anode materials for Li ion batteries? Computational studies on electronic properties and Li storage capability of Ti3C2 and Ti3C2X2 (X = F, OH) monolayer. J. Am. Chem. Soc. 2012, 134, 16909-16.
72. Wang, X.; Shen, X.; Gao, Y.; Wang, Z.; Yu, R.; Chen, L. Atomic-scale recognition of surface structure and intercalation mechanism of Ti3C2X. J. Am. Chem. Soc. 2015, 137, 2715-21.
73. Wen, J.; Zhang, X.; Gao, H. Role of the H-containing groups on the structural dynamics of Ti3C2Tx MXene. Physica. B. 2018, 537, 155-61.
74. Aierken, Y.; Sevik, C.; Gülseren, O.; Peeters, F. M.; Çakır, D. MXenes/graphene heterostructures for Li battery applications: a first principles study. J. Mater. Chem. A. 2018, 6, 2337-45.
75. Nair, A. K.; Da Silva, C. M.; Amon, C. H. Enhanced alkali-ion adsorption in strongly bonded two-dimensional TiS2/MoS2 van der Waals heterostructures. J. Phys. Chem. C. 2023, 127, 9541-53.
76. Ye, L.; Wu, S.; Wang, Z. Mechanical properties of two-dimensional materials (graphene, silicene and MoS2 monolayer) upon lithiation. J. Electron. Mater. 2020, 49, 5713-20.
77. Browne, S.; Waghmare, U. V.; Singh, A. Opportunities and challenges for 2D heterostructures in battery applications: a computational perspective. Nanotechnology 2022, 33, 272501.
78. Nyamdelger, S.; Ochirkhuyag, T.; Sangaa, D.; Odkhuu, D. First-principles prediction of a two-dimensional vanadium carbide (MXene) as the anode for lithium ion batteries. Phys. Chem. Chem. Phys. 2020, 22, 5807-18.
79. Li, X.; Wang, C.; Cao, Y.; Wang, G. Functional MXene materials: progress of their applications. Chem. Asian. J. 2018, 13, 2742-57.
80. Hantanasirisakul, K.; Gogotsi, Y. Electronic and Optical Properties of 2D transition metal carbides and nitrides (MXenes). Adv. Mater. 2018, 30, 1804779.
81. Xie, Y.; Naguib, M.; Mochalin, V. N.; et al. Role of surface structure on Li-ion energy storage capacity of two-dimensional transition-metal carbides. J. Am. Chem. Soc. 2014, 136, 6385-94.
82. Lu, M.; Han, W.; Li, H.; Zhang, W.; Zhang, B. There is plenty of space in the MXene layers: the confinement and fillings. J. Energy. Chem. 2020, 48, 344-63.
83. Tian, S.; Zhou, K.; Huang, C. Q.; Qian, C.; Gao, Z.; Liu, Y. Investigation and understanding of the mechanical properties of MXene by high-throughput computations and interpretable machine learning. Extreme. Mech. Lett. 2022, 57, 101921.
84. Kazemi, S. A.; Wang, Y. Super strong 2D titanium carbide MXene-based materials: a theoretical prediction. J. Phys. Condens. Matter. 2020, 32, 11LT01.
85. Persson, I.; Näslund, L. A.; Halim, J.; et al. On the organization and thermal behavior of functional groups on Ti3C2 MXene surfaces in vacuum. 2D. Mater. 2017, 5, 015002.
86. Tang, K.; Qi, W.; Wei, Y.; Ru, G.; Liu, W. High-throughput calculation of interlayer van der Waals forces validated with experimental measurements. Research 2022, 2022, 2022/9765121.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].