


Wilson disease revisited: is the gut a metabolically vulnerable target organ?
 0
0 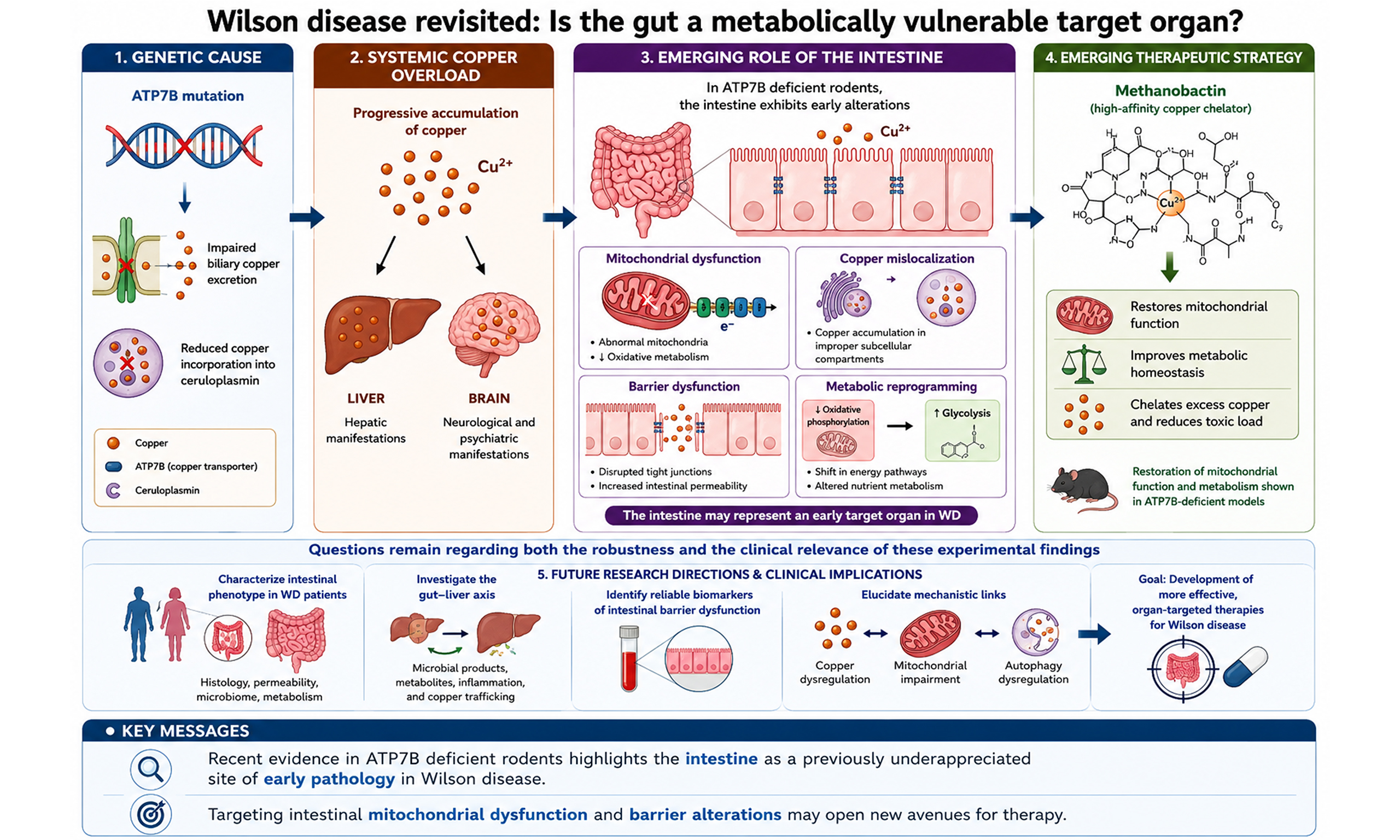
Wilson disease (WD) is a rare autosomal recessive disorder of copper metabolism caused by mutations in the ATP7B gene, which encodes a copper-transporting P-type ATPase predominantly expressed in hepatocytes. The defect impairs biliary copper excretion and copper incorporation into ceruloplasmin, resulting in progressive accumulation of toxic copper in the liver, brain, and other organs. The estimated WD prevalence is approximately 1 in 30,000 individuals[1].
COPPER HOMEOSTASIS
Copper is an essential trace element primarily obtained through the diet, with an average adult intake of approximately 1-3 mg/day. The intestine serves as the primary site of copper absorption and a key regulatory hub, controlling the uptake and delivery of dietary copper into the portal circulation[2]. Dietary copper, mainly in the Cu2+ (cupric) form, is absorbed in the duodenum and proximal small intestine. At the enterocyte brush border, Cu2+ is reduced to Cu+ (cuprous) by reductases and taken up primarily via the high-affinity copper transporter 1 (CTR1). Once in the enterocyte, copper is bound by intracellular chaperone proteins, including antioxidant protein 1 (ATOX1), copper chaperone for superoxide dismutase (CCS), and cytochrome c oxidase copper chaperone (COX17). ATOX1 delivers copper to the copper-transporting ATPase ATP7A, the dominant transporter, in the trans-Golgi network (TGN), where it is incorporated into cuproenzymes. CCS transfers copper to superoxide dismutase 1 (SOD1), and COX17 delivers copper to mitochondria for incorporation into cytochrome c oxidase. ATP7A continuously cycles between the TGN and the plasma membrane and, under conditions of copper excess, traffics to the basolateral membrane to facilitate copper export into the portal circulation. ATP7B is expressed at lower levels in enterocytes than in hepatocytes. In the intestine, ATP7B may contribute primarily to copper sequestration into vesicles and apical export into the intestinal lumen, particularly under conditions of copper excess.
The intestine acts as a key gatekeeper of systemic copper homeostasis through coordinated regulation of CTR1 and ATP7A. Under conditions of copper excess, CTR1 expression is downregulated, and ATP7A relocalizes to the basolateral membrane to enhance efflux and delivery to the liver; during copper deficiency, CTR1 expression increases, promoting copper uptake and intracellular retention in the TGN to prioritize enzyme synthesis. Once in portal circulation, copper is predominantly bound to albumin and to small peptides, such as histidine, for hepatic uptake.
Within hepatocytes, ATP7B has two central roles. First, in the TGN, ATP7B incorporates copper into apoceruloplasmin to generate holoceruloplasmin, which is subsequently secreted into the circulation. Second, when intracellular copper levels rise, ATP7B traffics to the canalicular membrane and facilitates copper excretion into bile. In bile, copper binds to bile salts, which helps prevent its reabsorption in the intestine. Approximately 95% of circulating copper is bound to ceruloplasmin, which delivers copper to peripheral tissues for enzymatic processes.
At the cellular level, copper serves as a catalytic cofactor for numerous cuproenzymes involved in mitochondrial respiration, oxidative phosphorylation, antioxidant defense, neurotransmitter synthesis, tyrosine metabolism, redox regulation, and extracellular matrix remodeling[3]. Both copper deficiency and excess are harmful: insufficient copper impairs cuproenzyme activity, whereas intracellular copper accumulation promotes oxidative stress, mitochondrial dysfunction, and cellular injury.
HEPATIC INJURY IN WD
WD may present at any age from early childhood to late adulthood[4,5]. Clinical manifestations are highly variable, but typically include hepatic involvement alongside neurological and psychiatric features. Hepatic manifestations span a broad spectrum, ranging from isolated mild elevations in transaminases to advanced cirrhosis with portal hypertension or acute liver failure.
Hepatic copper overload is widely considered the primary driver of liver injury in WD. In patients and animal models, including Atp7b-deficient mice and rats, mitochondrial copper accumulation is consistently observed[6]. Ultrastructural studies have shown that excess mitochondrial copper induces oxidative stress, disrupts cristae architecture, impairs bioenergetics, and may lead to mitochondrial DNA depletion[6,7]. These abnormalities occur early in the disease course and progressively worsen over time.
Collectively, these findings identify mitochondria as key targets of copper toxicity and implicate mitochondrial dysfunction as a central contributor to WD pathogenesis. This has prompted interest in mitochondria-targeted copper chelation as a potential therapeutic strategy.
However, the precise sequence of molecular events remains incompletely defined. In particular, it is unclear how excess copper is initially distributed within hepatocytes, and whether mitochondrial dysfunction arises from direct copper accumulation or secondary oxidative stress. The temporal relationship among mitochondrial damage, lysosomal impairment, and activation of compensatory pathways such as autophagy also remains poorly understood[8].
INTESTINAL INVOLVEMENT IN WD: CURRENT UNCERTAINTIES
WD has traditionally been viewed as a disorder primarily characterized by hepatic copper overload with secondary neurological involvement. Within this framework, the intestine has largely been considered a passive site of copper absorption rather than a target of copper-induced injury. However, recent work by Fontes et al. from the Hans Zischka research group (Munich, Germany) challenges this paradigm, suggesting that the intestinal epithelium may itself be an active and vulnerable site of pathology in WD[9].
Gastrointestinal symptoms occur in approximately 40% of patients and may precede diagnosis or be attributed to therapy-related side effects. Through analysis of intestinal biopsies from two WD patients, two animal models (WD rats and Atp7b⁻/⁻ mice), and ATP7B-deficient human intestinal epithelial cells,
Notably, intestinal pathology did not correlate with total intestinal copper concentrations. Instead, the authors propose that intracellular copper mislocalization and defective copper handling, rather than absolute copper load, drive cellular stress and barrier disruption.
In complementary studies, they further demonstrate that methanobactin therapy, a bacteria-derived chalkophore with high copper affinity, restores intestinal barrier integrity, improves mitochondrial ultrastructure, and reduces inflammatory changes in WD animal models and ATP7B-deficient human enterocytes[9]. These findings support copper as a driver, rather than a bystander, of the intestinal phenotype.
Taken together, Fontes et al. propose a model in which ATP7B deficiency leads to early intestinal alterations, including marked mitochondrial abnormalities and reduced oxidative metabolism, subcellular copper mislocalization rather than absolute copper accumulation, epithelial barrier dysfunction, and metabolic reprogramming[9]. They highlight the intestine as a potentially important site of early pathology and a candidate target for therapeutic intervention.
However, questions remain regarding both the robustness and clinical relevance of these experimental findings. First, the human data are limited and confounded: the only two WD patients who underwent intestinal biopsy had comorbid inflammatory conditions, namely Crohn’s disease and suspected coeliac disease, both of which can independently cause villous atrophy, crypt distortion, and mucosal inflammation. Therefore, it remains unclear whether the observed lesions are specific to WD. Second, the assumption that subcellular copper “mislocalization” drives cellular stress and barrier disruption should be interpreted with caution, as it is based on crude fractionation approaches and lacks direct evidence of organelle-specific copper redistribution. A more robust approach would combine quantitative subcellular fractionation (e.g., mitochondria, lysosomes, ER/Golgi, and cytosol) with imaging-based elemental mapping to directly visualize intracellular copper distribution. Together, these limitations preclude definitive conclusions regarding causality and translational relevance.
Finally, the observation that intestinal alterations may precede overt hepatic disease in rats raises broader questions about their potential contribution to the development of liver disease. Studies of intestinal microbial responses to different copper ion (Cu2+) concentrations have shown that in healthy children, low-dose physiological copper exposure (e.g., 2 mg/L) increases the abundance of beneficial bacteria such as Lactobacillus and Lactococcus, activates antioxidant and detoxification pathways, and supports intestinal homeostasis[10]. In contrast, exposure above a certain threshold (≥ 4 mg/L) disrupts microbial balance, reduces the abundance of beneficial taxa, promotes enrichment of potentially pathogenic bacteria, and suppresses the expression of genes involved in metabolism and detoxification, ultimately leading to intestinal barrier impairment and inflammatory responses[10]. Although studies on gut dysbiosis in WD remain limited, reduced microbial diversity and a shift toward a more pro-inflammatory microbial profile have been reported in WD patients[11]. Several plausible mechanisms may underlie microbiome alterations, including the antimicrobial effects of excess copper[2], changes in bile acid metabolism secondary to liver disease[11], and intestinal barrier impairment increasing immune activation[2]. In turn, gut dysbiosis may exacerbate disease progression through increased translocation of microbial products and modulation of inflammatory pathways along the gut-liver axis[12]. However, whether dysbiosis is a driver of intestinal barrier dysfunction and liver injury, or instead represents a secondary consequence of hepatic pathology, remains unresolved. Future studies should therefore assess intestinal permeability and the translocation of microbial products (e.g., endotoxins) into the portal circulation in WD patients, and examine their correlation with hepatic copper levels and disease severity.
THERAPEUTIC STRATEGIES FOR WD
Current pharmacological approaches
Current treatment approaches aim to reduce systemic copper burden either by limiting intestinal copper absorption through zinc-induced metallothionein-mediated sequestration of copper within enterocytes or by enhancing urinary copper excretion using chelators such as D-penicillamine and trientine[4]. However, hepatic copper concentrations frequently remain elevated despite long-term therapy[13]. Moreover, whether complete removal of excess hepatic copper would constitute a true cure for WD remains uncertain. Advances in mechanistic understanding of WD, particularly regarding mitochondrial injury, may inform the development of more targeted therapies.
Methanobactin: proof-of-concept, not an established clinical therapy
Preclinical studies have demonstrated that methanobactin, a high-affinity bacterial copper chelator, rapidly depletes mitochondrial copper, reverses liver injury, and improves survival in ATP7B-deficient models, supporting its potential as a novel therapeutic strategy for WD[14-17]. Fontes et al. further demonstrated that methanobactin therapy restores intestinal alterations and improves cellular metabolism in WD animal models and ATP7B-deficient human enterocytes[9].
However, given the current lack of clinical trial data, translation to clinical use in humans requires further investigation. Several important challenges remain, including peptide stability, delivery, pharmacokinetic profile and tissue distribution, safety, particularly the risk of off-target depletion of essential metals, optimal dosing, and long-term effects.
Methanobactin (ARBM101) is now part of the pipeline of a pharmaceutical company (Arbormed, https://www.arbormed.com/en/).
FUTURE DIRECTIONS
Several key questions warrant investigation
First, intestinal biopsies from treatment-naïve WD patients without confounding gastrointestinal disease are essential to validate the proposed intestinal phenotype in humans. Longitudinal studies in presymptomatic individuals and pediatric populations could help determine whether intestinal barrier dysfunction precedes hepatic injury or evolves in parallel.
Second, deeper mechanistic exploration of the gut–liver axis, including the roles of microbiota composition, microbial metabolites, and immune signaling, may clarify how intestinal pathology influences systemic disease progression. Biomarkers reflecting intestinal permeability or epithelial metabolic function could also prove valuable for disease monitoring and therapeutic stratification.
Finally, the precise mechanisms underlying the beneficial effects of methanobactin remain incompletely defined. It is unclear whether the observed improvements result solely from copper depletion or also involve modulation of autophagic flux, redox balance, or broader metabolic pathways. Advanced experimental models, including patient-derived intestinal organoids, high-resolution copper imaging techniques, and detailed dissection of copper-mitochondria–autophagy interactions, will be required before intestine-targeted therapeutic strategies can be rationally integrated into WD management.
CONCLUDING REMARKS
The study by Fontes et al. expands the conceptual framework of WD from a predominantly hepatocentric disorder to a multisystem disease that includes early intestinal involvement[9]. Across species and experimental systems, ATP7B deficiency is associated with intestinal inflammation, impaired barrier integrity, mitochondrial dysfunction, and altered metabolism. These findings underscore defective copper trafficking and compartmentalization, rather than simple copper overload, as potential drivers of tissue injury. Preclinical studies with methanobactin demonstrate that targeted copper chelation can reverse mitochondrial and barrier abnormalities, but this approach remains experimental and mechanistically incompletely defined. The clinical relevance of these findings derived from animal models remains uncertain.
Overall, this study should be regarded as a hypothesis-generating framework rather than a basis for immediate changes in clinical practice. Its principal contribution lies in reframing WD as a disorder of systemic mitochondrial vulnerability to copper toxicity and in opening new avenues for mechanistically informed therapeutic development.
DECLARATIONS
Authors’ contributions
Involved in the conception and design of the manuscript: Debray D, Poujois A
Drafted the manuscript: Debray D
Critically revised the manuscript for intellectual content: Poujois A
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (OpenAI; GPT-5.5, released 2026-04-23) was used solely for language editing and the preparation of the Graphical Abstract. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
None.
Conflicts of interest
Dominique Debray is an adviser for Orphalan. Dominique Debray has previously co-authored publications with Piotr Socha, who is an author of the article discussed in this commentary. This relationship may be perceived as a potential conflict of interest; however, the authors affirm that it did not influence the content or interpretation of this commentary. Aurelia Poujois is an adviser for Orphalan, Alexion, Vivet, and Univar, and has received institutional grants from Orphalan, Alexion, and AddMedica.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Fang S, Furegato M, Azzi J, Couchonnal-Bedoya E, Debray D. Epidemiology and economic burden of Wilson disease in France: a nationwide population-based study. J Inherit Metab Dis. 2025;48:e12822.
2. Lu F, Wang X, Xue X, et al. Multifaceted role of copper homeostasis in gut health: from molecular mechanisms to therapeutic interventions. Cells. 2026;15:545.
3. Chen J, Jiang Y, Shi H, Peng Y, Fan X, Li C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflugers Arch. 2020;472:1415-29.
4. European Association for the Study of the Liver. EASL-ERN Clinical Practice Guidelines on Wilson’s disease. J Hepatol. 2025;82:690-728.
5. Nilles C, Obadia MA, Sobesky R, et al. Diagnosis and outcomes of late-onset Wilson’s disease: a national registry-based study. Mov Disord. 2023;38:321-32.
6. Amin R, Medici V, Fausak ED, et al. Mitochondrial dysfunction in Wilson disease: a systematic review and meta-analysis across human and animal models. Front Mol Biosci. 2025;12:1712573.
7. Nagasaka H, Inoue I, Inui A, et al. Relationship between oxidative stress and antioxidant systems in the liver of patients with Wilson disease: hepatic manifestation in Wilson disease as a consequence of augmented oxidative stress. Pediatr Res. 2006;60:472-7.
8. Polishchuk EV, Merolla A, Lichtmannegger J, et al. Activation of autophagy, observed in liver tissues from patients with Wilson disease and from ATP7B-deficient animals, protects hepatocytes from copper-induced apoptosis. Gastroenterology. 2019;156:1173-89.e5.
9. Fontes A, Pierson H, Bierła JB, et al. Copper impairs the intestinal barrier integrity in Wilson disease. Metabolism. 2024;158:155973.
10. Zhao L, Li X, Wang Y, et al. Resistance role of Lactobacillus sp. and Lactococcus sp. to copper ions in healthy children’s intestinal microorganisms. J Hazard Mater. 2024;469:134059.
11. Wei T, Qian N, Wang H, et al. Wilson’s disease-associated gut dysbiosis: novel insights into microbial functional alterations, virulence changes, and resistance markers. Front Microbiol. 2025;16:1714276.
12. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: pathophysiological basis for therapy. J Hepatol. 2020;72:558-77.
13. Gibbs K, Walshe JM. Liver copper concentration in Wilson’s disease: effect of treatment with ‘anti-copper’ agents. J Gastroenterol Hepatol. 1990;5:420-4.
14. Lynderup EM, Vendelbo MH, Kirk FT, et al. Methanobactin rapidly facilitates biliary copper excretion in a Wilson disease rat model visualised by 64Cu PET/MRI. Br J Pharmacol. 2026;183:268-79.
15. Engler J, Kim EJ, Kim D, et al. Rapid transcellular hepatic copper depletion by ARBM-101 rescues severe liver damage in Wilson disease rodents. Biomed Pharmacother. 2025;193:118867.
16. Einer C, Munk DE, Park E, et al. ARBM101 (Methanobactin SB2) drains excess liver copper via biliary excretion in Wilson’s disease rats. Gastroenterology. 2023;165:187-200.e7.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].