





Immunotherapy resistance and strategies in malignant pleural mesothelioma
 0
0 Abstract
Malignant pleural mesothelioma (MPM) remains one of the most aggressive thoracic malignancies, characterized by profound resistance to conventional modalities such as surgery, chemotherapy, and radiotherapy, resulting in persistently poor survival outcomes. The advent of immune checkpoint inhibitors (ICIs) has fundamentally reshaped the therapeutic landscape of MPM. Notably, dual programmed cell death protein 1 (PD-1)/cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) blockade has demonstrated superior efficacy over monotherapy in multiple phase I/II trials and has been established as a novel first-line standard of care. Nevertheless, the high incidence of resistance continues to pose a major clinical challenge. This therapeutic bottleneck is largely attributed to the unique biology of MPM, including a profoundly immunosuppressive tumor microenvironment, aberrantly activated signaling pathways, and complex metabolic reprogramming, which together form a multilayered defense network against immune attack. In response to this intricate resistance architecture, recent research efforts have increasingly focused on the development of precision combination strategies. By rationally integrating ICIs with anti-angiogenic agents, chemotherapy, metabolic modulators, and next-generation cellular immunotherapies [e.g., chimeric antigen receptor T cells (CAR-T), chimeric antigen receptor-natural killer (CAR-NK)], these approaches aim to dismantle immune evasion barriers and reinvigorate antitumor immunity. Concurrently, the discovery of novel biomarkers and their integration with multi-omics data are enabling more precise patient stratification, signaling the advent of an era of personalized immunotherapy for MPM. This review provides a systematic synthesis of the latest clinical advances and fundamental breakthroughs in MPM immunotherapy, with a particular focus on dissecting the multifactorial mechanisms underlying therapeutic resistance. Its core contribution lies in constructing a forward-looking framework for next-generation treatment strategies. It critically evaluates the translational potential of emerging approaches, including arginine deprivation therapy for argininosuccinate synthase 1 (ASS1)-deficient tumors, CAR-T cells, T-cell receptor fusion constructs, and oncolytic virotherapy. By integrating these innovative modalities with biomarker-guided patient selection, this review delineates a roadmap for transitioning MPM management from empirical therapy toward precision immuno-oncology, with the ultimate goal of achieving durable disease control in this challenging malignancy.
Keywords
INTRODUCTION
Malignant pleural mesothelioma (MPM) is an aggressive and lethal malignancy arising from the mesothelial cells of the pleural cavity, most commonly linked to asbestos exposure[1]. Despite advances in understanding its pathogenesis, the overall prognosis remains dismal, with median overall survival (OS) reportedly limited to 12-18 months under conventional therapies[2]. The global incidence, though declining in several developed countries due to asbestos regulation, continues to rise in developing regions where industrial use persists, reflecting the disease’s long latency period of 30-50 years[3,4].
For nearly two decades, the standard first-line regimen for unresectable MPM consisted of platinum-based chemotherapy combined with pemetrexed, as established in the pivotal 2003 Vogelzang trial. However, despite modest improvements in progression-free survival (PFS) and symptom control, long-term outcomes plateaued, and second-line options offered minimal benefit. Multimodal approaches integrating surgery and radiotherapy were limited to selected patients, with high recurrence rates and substantial morbidity[5,6].
The emergence of immune checkpoint inhibitors (ICIs) has substantially reshaped the therapeutic paradigm for MPM, although their clinical benefit remains heterogeneous across patient subgroups. The CheckMate 743 phase III trial represented a turning point, demonstrating a significant OS benefit for nivolumab plus ipilimumab over standard chemotherapy [median OS 18.1 vs. 14.1 months; hazard ratio (HR) ≈ 0.73], particularly among patients with non-epithelioid histology. Subsequent studies such as KEYNOTE-483 (NCT02784171) and BEAT-meso (NCT03762018) confirmed the value of immunotherapy either as monotherapy or in combination with chemotherapy and anti-angiogenic agents, leading to the formal recognition of dual ICI therapy as a standard first-line treatment for unresectable MPM[7-9].
Nevertheless, not all patients benefit from ICIs, and a considerable fraction exhibits primary or acquired resistance. Tumor heterogeneity, a profoundly immunosuppressive microenvironment, and a low mutational burden collectively contribute to objective response rates (ORRs) - typically < 25% for monotherapy. Moreover, the absence of validated predictive biomarkers and the emergence of immune-related adverse events (irAEs) complicate clinical management[10-12].
In this context, research in MPM has rapidly shifted from establishing immunotherapy efficacy toward understanding and overcoming resistance. Current investigations aim to elucidate the molecular and cellular determinants of immune evasion, identify predictive biomarkers, and design rational combination strategies that enhance immune responsiveness. This review provides a comprehensive overview of the immunopathogenesis of MPM, clinical advances in immunotherapy, mechanisms of therapeutic resistance, and emerging strategies - including metabolic targeting, cellular immunotherapies, and oncolytic platforms - that are poised to redefine the management of this challenging malignancy[13-16].
IMMUNOPATHOGENESIS AND TUMOR MICROENVIRONMENT: MOLECULAR AND IMMUNOLOGIC BASIS OF MESOTHELIOMA
Asbestos exposure, responsible for nearly 80% of MPM, initiates a decades-long cascade of chronic inflammation and oxidative injury[17]. Biopersistent fibers lodged in the pleura trigger frustrated phagocytosis, generating persistent reactive oxygen species (ROS)/reactive nitrogen species (RNS), DNA damage, chromosomal instability, and release of pro-tumorigenic cytokines [tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β, transforming growth factor-β (TGF-β)][18]. Over a 30-50-year latency, these genotoxic and inflammatory pressures drive malignant transformation. Genomically, MPM is dominated by loss-of-function alterations rather than classical oncogene activation. BRCA1-associated protein 1 (BAP1) loss (~60%) disrupts chromatin regulation and DNA repair, producing an “inflamed” transcriptional phenotype with enhanced interferon signaling and greater ICI sensitivity in some studies (e.g., NCT02899299, NCT01773655)[19]. Cyclin-dependent kinase inhibitor 2A (CDKN2A) deletion (~50%) inactivates the retinoblastoma protein (Rb)/tumor protein p53 (p53) pathways, correlates with non-epithelioid histology and poor prognosis, and may impair antigen presentation[20]. Neurofibromin 2 (NF2) alterations (~40%) activate the Hippo–Yes-associated protein (YAP) axis, promoting proliferation, epithelial-mesenchymal transition (EMT), and immunosuppressive cytokines [IL-6, C-X-C motif chemokine ligand 5 (CXCL5)], along with programmed death-ligand 1 (PD-L1) and vascular endothelial growth factor (VEGF) upregulation; transcriptional enhanced associate domain (TEAD) inhibitors (e.g., IK-930, NCT05228015) are under clinical evaluation in NF2-mutant tumors. TEAD inhibition specifically addresses the Hippo pathway dysregulation discussed in Section MECHANISTIC INSIGHTS INTO THERAPEUTIC ACTION, restoring immune susceptibility in NF2-mutant tumors by modulating the immunosuppressive cytokine milieu and potentially enhancing susceptibility to ICI therapy. Sarcomatoid MPM displays a distinct immunobiological profile characterized by elevated PD-L1 expression, enhanced interferon-related transcriptional programs, and increased immune infiltration compared with epithelioid tumors. Notably, subgroup analyses from CheckMate 743 demonstrated that patients with non-epithelioid histology derived a disproportionately greater survival benefit from dual immune checkpoint blockade, suggesting that sarcomatoid tumors may represent an inflamed yet immunologically restrained subtype particularly amenable to checkpoint-based strategies. Recent multiplex immunofluorescence analyses have further elucidated the molecular basis of this heightened sensitivity. A study using an 8-marker panel revealed that sarcomatoid tumors exhibit significantly higher proportions of cells expressing cyclooxygenase-2 (COX2) (92.6% vs. 80.9%, P < 0.001), transforming growth factor-beta (TGFB) (86.3% vs. 65.0%, P < 0.001), T-cell immunoreceptor with Ig and ITIM domains (TIGIT, 91.7% vs. 67.4%, P < 0.001), and T-cell immunoglobulin and mucin domain-containing protein 3 (TIM3) (59.7% vs. 20.6%, P < 0.001) compared to epithelioid tumors. These findings provide a mechanistic rationale for the enhanced responsiveness of sarcomatoid MPM to dual checkpoint blockade and identify potential combinatorial targets for this aggressive subtype[21,22].
These genomic aberrations converge with a profoundly immunosuppressive tumor microenvironment (TME). MPM lesions are enriched in M2-polarized tumor-associated macrophages (TAMs) producing IL-10, TGF-β, and VEGF, suppressing dendritic cell (DC) maturation and correlating with poor survival[23]. Regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) expand in response to chronic inflammation, dampening cytotoxic T-cell proliferation via IL-10, TGF-β, and arginase-1–mediated L-arginine depletion - a mechanism reinforced by tumor-intrinsic argininosuccinate synthase 1 (ASS1) deficiency[24,25]. VEGF-driven angiogenesis and hypoxia further impair T-cell trafficking while promoting Treg/MDSC recruitment, providing the rationale for anti-angiogenic and ICI combinations validated in BEAT-meso (NCT03762018)[25,26]. MPM additionally expresses multiple checkpoint and inhibitory molecules [PD-L1, Galectin-9, B7 Homolog 3 (B7-H3)], while TGF-β signaling suppresses major histocompatibility complex class I (MHC-I) and facilitates fibroblast activation; such pathways contribute to resistance to programmed cell death protein 1 (PD-1)/PD-L1 blockade and underpin ongoing TGF-β–targeted trials [e.g., TGF-β2-targeting antisense oligonucleotide (OT-101) + pembrolizumab, NCT05425576][26-29].
Together, asbestos-induced oxidative stress, tumor-suppressor loss, and a structurally and functionally immunosuppressive TME form a continuous pathogenic axis that drives tumor initiation and immune escape. These intertwined mechanisms constitute the biological framework guiding current immunotherapy strategies and next-generation approaches aimed at metabolic targeting, TME reprogramming, and pathway-specific intervention[30,31].
CLINICAL DEVELOPMENT OF ICIS IN MPM
The clinical development of ICIs in MPM has evolved from early exploratory phase II studies to multiple phase III randomized controlled trials, establishing immunotherapy as a new therapeutic pillar beyond platinum–pemetrexed chemotherapy. Early evidence came from the MAPS2 trial (NCT02716272), which compared nivolumab alone vs. nivolumab plus ipilimumab in relapsed MPM[32]. The 12-week disease control rate (DCR) reached 40% and 52%, respectively, with a median OS of 11.9 and 15.9 months - demonstrating manageable safety but increased irAEs in the dual-checkpoint cohort. In contrast, the PROMISE-meso trial (NCT02991482) compared pembrolizumab with investigator’s-choice chemotherapy and reported a higher ORR (22% vs. 6%) but no significant OS or PFS benefit, underscoring the limitation of PD-1 monotherapy[33,34].
Definitive phase III evidence came from the CheckMate 743 trial, which compared nivolumab plus ipilimumab against platinum–pemetrexed in the first-line setting. The dual-immunotherapy regimen achieved a median OS of 18.1 vs. 14.1 months (HR ≈ 0.73), with durable 3-year survival rates of 23.2% vs. 15.4%[35]. Notably, the survival advantage was most pronounced in non-epithelioid histology (OS 18.1 vs. 8.8 months, HR ≈ 0.46), reflecting the biologic heterogeneity of immune responsiveness in MPM. These findings led to the U.S. Food and Drug Administration (FDA) approval of nivolumab–ipilimumab as first-line therapy in 2020, marking a paradigm shift in disease management[36-38].
More recently, combination chemoimmunotherapy has emerged as a potential refinement. The KEYNOTE-483 trial (IND227) evaluated pembrolizumab with platinum–pemetrexed vs. chemotherapy alone and demonstrated a median OS of 17.3 vs. 16.1 months (HR = 0.79), supporting regulatory recognition in 2024[8]. The ongoing DREAM3R (NCT04334759) and BEAT-meso trials (NCT03762018) are further investigating immunotherapy integrated with chemotherapy and anti-angiogenic agents[39]. Despite these advances, response rates remain modest, long-term survival is limited to a subset of patients, and immune-related toxicities present new management challenges[40]. Collectively, these data highlight that while ICIs have redefined the therapeutic landscape of MPM, overcoming primary and acquired resistance remains the next frontier[41,42].
MECHANISTIC INSIGHTS INTO THERAPEUTIC ACTION
The limited efficacy and heterogeneous clinical response to ICIs in MPM reflect a complex interplay of immune, stromal, metabolic, and genomic mechanisms that collectively drive both primary refractoriness and acquired resistance during treatment. A defining pathological feature of MPM is its chronically inflamed yet immunologically “restricted” TME, where effector T-cell recruitment, activation, and long-term persistence are impeded by multiple layers of suppressive signaling[43]. At the cellular level, enrichment of Tregs constitutes a major barrier to antitumor immunity[44,45]. These cells accumulate in pleural tumor nodules and effusions, maintained by continuous exposure to IL-6, TGF-β, and tumor-derived prostaglandins[46]. Once present, Tregs inhibit DC maturation, outcompete CD8+ T cells for IL-2, and deliver direct suppressive signals via CTLA-4–mediated trans-endocytosis of CD80/86 from antigen-presenting cells. This constellation of effects results in attenuated priming and impaired expansion of tumor-reactive T-cell clones[47-50].
MDSCs further reinforce this tolerance landscape. Driven by persistent asbestos-induced inflammation, high mobility group box 1 protein (HMGB1) release, and tumor-secreted granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-8, MDSCs accumulate in both systemic circulation and the pleural cavity[50]. Through high-level expression of arginase-1, inducible nitric oxide synthase (iNOS), and ROS–generating enzymes, MDSCs inhibit T-cell receptor (TCR) ζ-chain expression, curtail antigen-specific proliferation, and restrict T-cell infiltration via chemokine remodeling[51]. Importantly, this immunosuppressive program converges with a core metabolic vulnerability of MPM: deficiency of ASS1. Loss of ASS1 - especially prevalent in non-epithelioid tumors - renders MPM highly dependent on exogenous arginine. As MDSCs consume arginine at a high rate, effector lymphocytes are metabolically starved, creating a competitive disadvantage that profoundly limits their activation threshold and cytotoxic capacity. These processes act in concert to establish a baseline state of ICI resistance even before treatment begins[52-55].
In addition to cellular and metabolic obstacles, intrinsic genomic features shape ICI responsiveness. MPM typically carries a low tumor mutational burden (TMB), limiting neoantigen availability and reducing the probability of pre-existing T-cell clones capable of recognizing tumor antigens[56]. Loss-of-function alterations in BAP1, NF2, and components of the Hippo–YAP signaling pathway modulate interferon signaling, antigen presentation, and cytokine release. While some BAP1-deficient tumors exhibit increased immune infiltration, a significant proportion display impaired type I interferon responses and reduced MHC class I expression, enabling immune evasion and promoting the emergence of antigen-loss variants during ICI therapy[57,58]. NF2 mutation, ASS1 deficiency, and BAP1 loss should not be viewed as isolated genomic events but as components of an interconnected metabolic–immune circuitry. NF2 inactivation drives Hippo pathway dysregulation and mammalian target of rapamycin (mTOR) activation, thereby increasing anabolic demand and nutrient dependence. In the setting of ASS1 deficiency, enforced arginine auxotrophy imposes metabolic rigidity, coupling proliferative signaling to nutrient vulnerability. This convergence may simultaneously sensitize tumors to arginine deprivation and intensify metabolic competition within the TME, with potential consequences for T-cell fitness and immune responsiveness. By contrast, BAP1 loss confers context-dependent immunologic effects. While frequently associated with interferon signaling, increased CD8+ infiltration, and elevated PD-L1 expression, BAP1 deficiency may also facilitate compensatory immune-evasive pathways and metabolic rewiring. Its impact on immune checkpoint blockade is therefore shaped by histologic context and co-occurring genomic alterations, underscoring the need to interpret BAP1 loss within a broader immunogenomic framework rather than as a standalone predictive marker. These interconnected vulnerabilities provide a rationale for mechanism-guided combinatorial strategies in MPM[59-63].
The stromal architecture of MPM further compounds therapeutic resistance. Dense fibrous stroma, abundant cancer-associated fibroblasts (CAFs), and dysfunctional neovasculature driven by VEGF impede lymphocyte trafficking and generate hypoxic, lactate-rich niches that destabilize T-cell mitochondrial metabolism[64]. VEGF-mediated inhibition of DC maturation also contributes to insufficient antigen priming, reinforcing a feedback loop of ineffective T-cell–mediated antitumor surveillance. These stromal barriers are particularly relevant in patients receiving ICIs, where the absence of adequate lymphocyte infiltration precludes the establishment of sustained antitumor immunity[65-68].
Cytokine-mediated mechanisms also contribute to treatment failure. High baseline levels of TGF-β, IL-10, and IL-1β suppress T-cell recruitment and effector differentiation, promote EMT, and remodel fibroblast and macrophage phenotypes toward immunosuppressive states[69,70]. During ICI therapy, compensatory upregulation of alternative checkpoints, such as T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), lymphocyte-activation gene-3 (LAG-3), TIGIT, and V-domain immunoglobulin (Ig) suppressor of T cell activation (VISTA), further contributes to T-cell exhaustion and loss of cytotoxic function. These adaptive pathways represent hallmark signatures of acquired resistance across multiple clinical cohorts[71-75].
Collectively, these intertwined cellular, stromal, metabolic, genomic, and cytokine-driven factors establish a multilayered resistance architecture unique to MPM. Understanding this complex ecosystem is essential for guiding rational combination strategies - particularly those integrating ICIs with metabolic modulation, anti-angiogenic agents, TGF-β inhibition, cellular immunotherapies, and genetically informed targeted agents. Overcoming resistance in MPM requires simultaneous disruption of multiple suppressive circuits rather than reliance on any single therapeutic axis, reinforcing the need for precision, multimodal immuno-oncologic approaches[76-79].
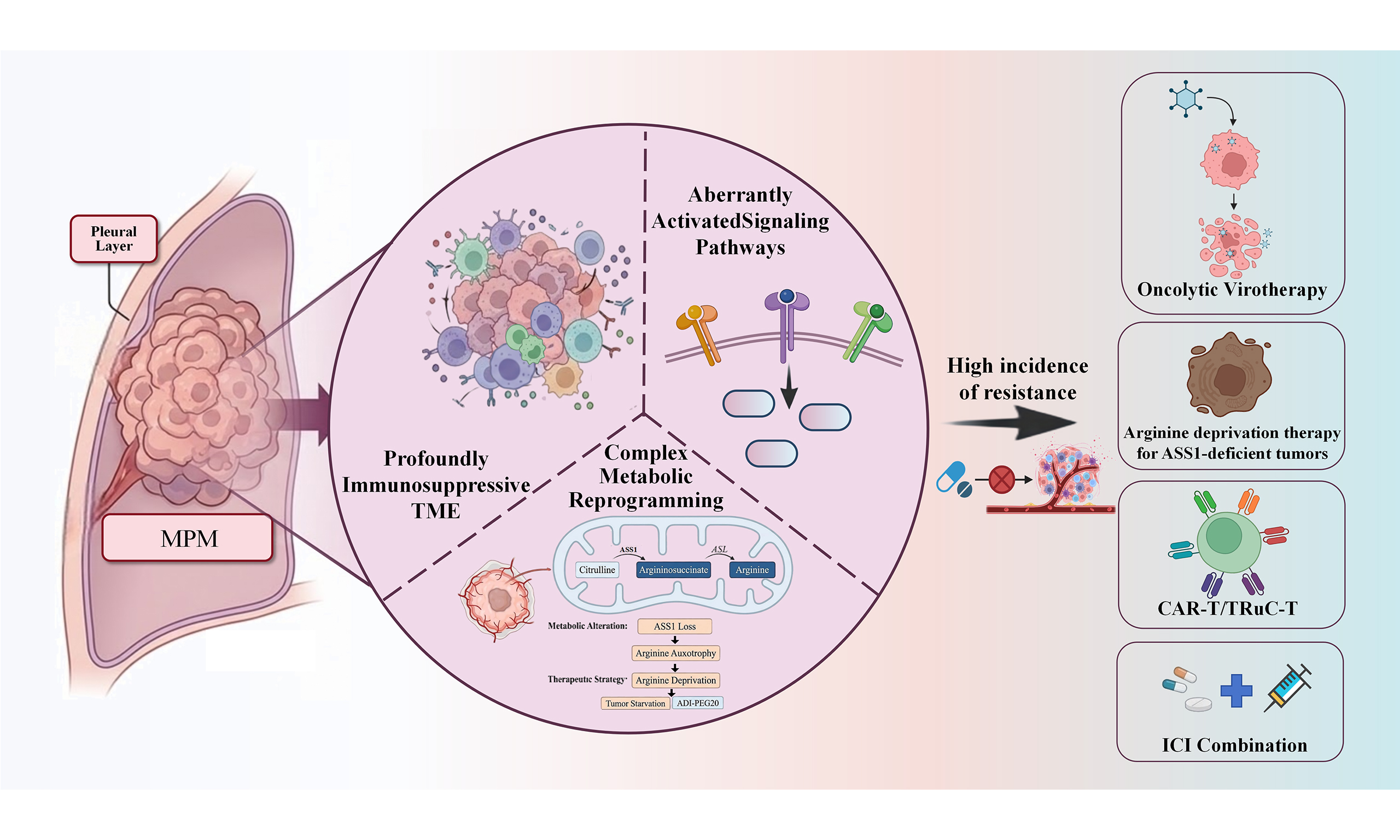
As summarized in Figure 1, these convergent pathways - including ASS1-mediated metabolic arginine dependency, Treg and MDSC accumulation, fibroblast-driven stromal stiffening, hypoxia, and alternative immune checkpoint upregulation - form a multilayered suppressive network that restrains cytotoxic T-cell infiltration and effector function.
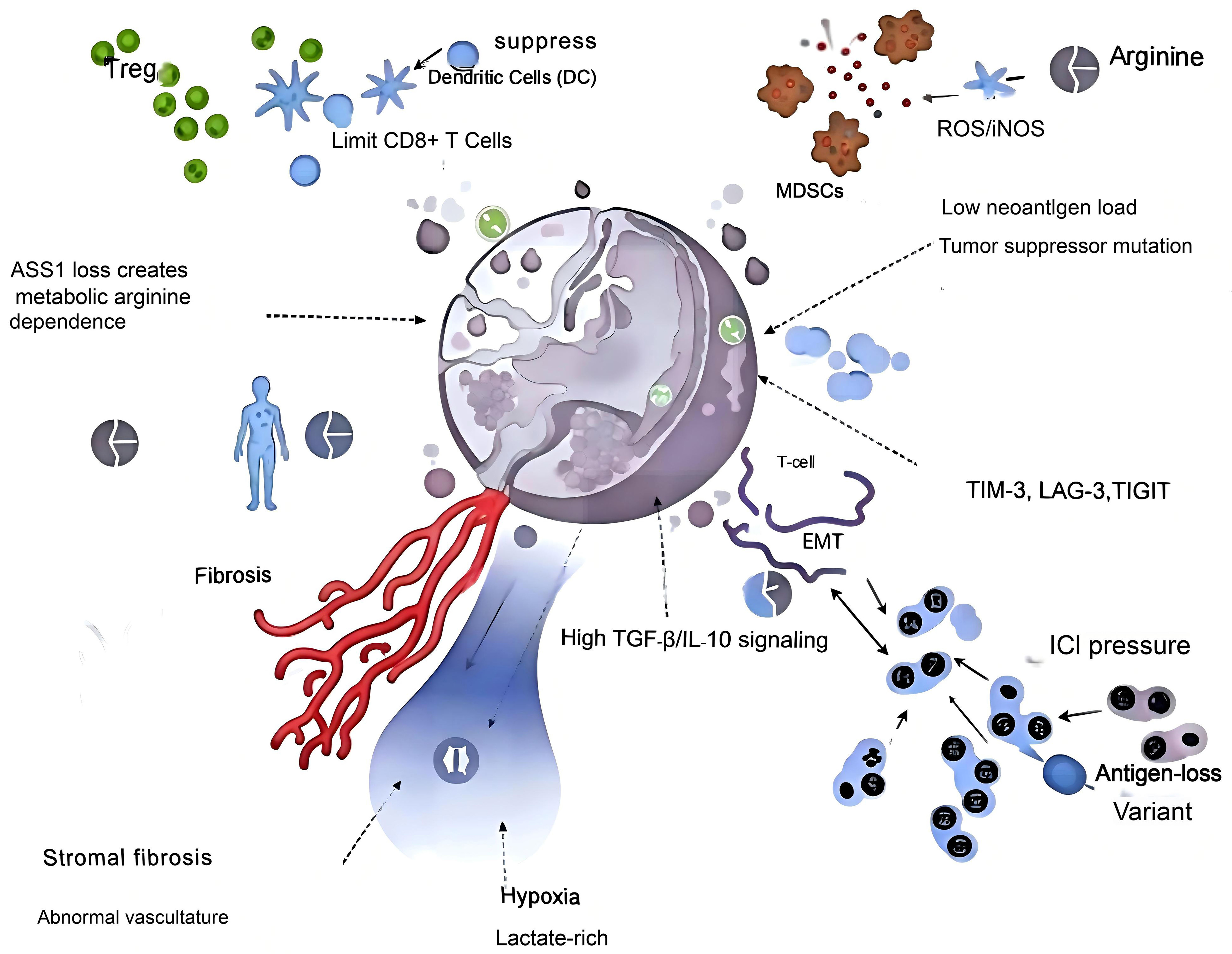
Figure 1. Key drivers of T cell–mediated immune resistance in MPM. MPM: Malignant pleural mesothelioma; Treg: regulatory T cell; DC: dendritic cell; CD8+ T cell: cytotoxic T lymphocyte; MDSCs: myeloid-derived suppressor cells; ROS: reactive oxygen species; iNOS: inducible nitric oxide synthase; ASS1: argininosuccinate synthase 1; TGF-β: transforming growth factor beta; IL-10: interleukin-10; EMT: epithelial-mesenchymal transition; TIM-3: T-cell immunoglobulin and mucin-domain containing-3; LAG-3: lymphocyte-activation gene-3; TIGIT: T-cell immunoreceptor with Ig and ITIM domains; ICI: immune checkpoint inhibitor.
STRATEGIES TO OVERCOME IMMUNOTHERAPY RESISTANCE IN MPM
The multifaceted resistance mechanisms delineated in this chapter - ranging from cellular suppressors (Tregs, MDSCs) and metabolic constraints (ASS1 deficiency) to genomic alterations (BAP1, NF2, CDKN2A) and stromal barriers (CAFs, VEGF-driven angiogenesis) - provide a rational framework for designing intervention strategies. Rather than targeting these obstacles in isolation, effective therapeutic combinations must simultaneously disrupt multiple layers of this suppressive network[80-83]. This section systematically maps each major resistance mechanism to its corresponding counter-strategy, establishing a mechanistic closed loop between the vulnerabilities characterized in Section MECHANISTIC INSIGHTS INTO THERAPEUTIC ACTION and their therapeutic targeting in Section STRATEGIES TO OVERCOME IMMUNOTHERAPY RESISTANCE IN MPM. Given the multifactorial nature of primary and acquired resistance in MPM, rational therapeutic strategies have increasingly focused on reprogramming the immunosuppressive TME, restoring antigen presentation, enhancing T-cell infiltration, and targeting tumor-specific metabolic and signaling dependencies. To systematically illustrate the key therapeutic directions that have emerged, Table 1 summarizes the representative clinical trials conducted from 2020-2025 exploring different approaches to overcoming immunotherapy resistance in MPM. This chapter provides a comprehensive synthesis of the most promising strategies designed to dismantle these resistance mechanisms, with the ultimate goal of restoring and sustaining effective anti-tumor immunity in MPM[32,84-87]. These strategies include anti-angiogenic approaches, chemo-immunotherapy combinations, metabolic modulation (e.g., arginine deprivation), TGF-β blockade, chimeric antigen receptor T cell (CAR-T)-based cellular immunotherapy, oncolytic viral therapy, dendritic-cell vaccination, and epigenetic/Hippo-pathway targeting. Figure 2 provides an illustrated overview of the immunosuppressive network in MPM and how each intervention point disrupts immune escape mechanisms.
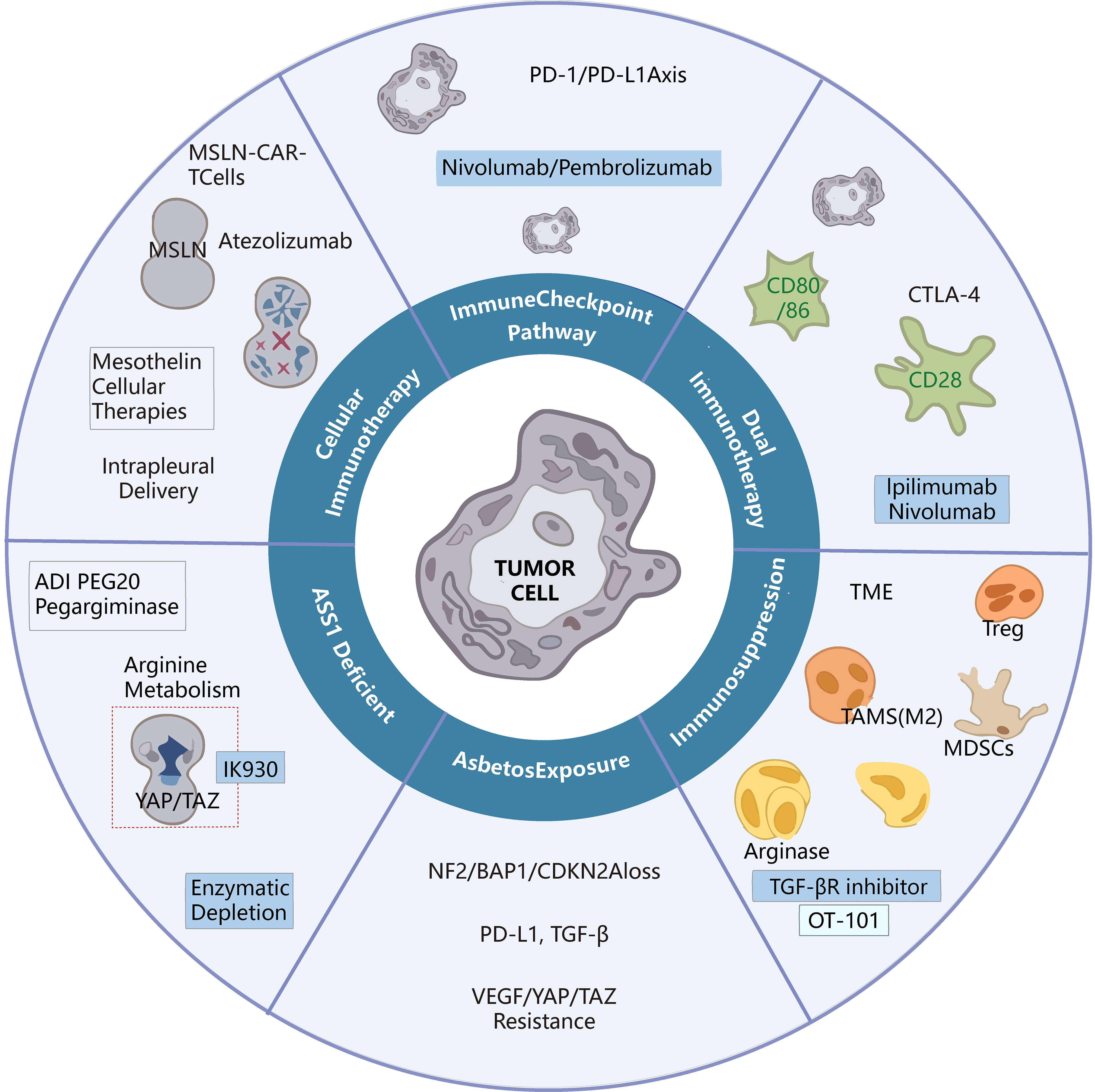
Figure 2. Strategies to overcome immunotherapy resistance in MPM. MPM: Malignant pleural mesothelioma; MSLN: mesothelin; CAR-T: chimeric antigen receptor T cell; Atezolizumab: anti-PD-L1 monoclonal antibody; PD-1: programmed cell death protein 1; Nivolumab: anti-PD-1 monoclonal antibody; Pembrolizumab: anti-PD-1 monoclonal antibody; CTLA-4: cytotoxic T-lymphocyte-associated protein 4; Ipilimumab: anti-CTLA-4 monoclonal antibody; TME: tumor microenvironment; Treg: regulatory T cell; TAMs: tumor-associated macrophages; MDSCs: myeloid-derived suppressor cells; TGF-βR: transforming growth factor-β receptor; OT-101: TGF-β2-targeting antisense oligonucleotide; NF2: neurofibromin 2; BAP1: BRCA1-associated protein 1; CDKN2A: cyclin-dependent kinase inhibitor 2A; PD-L1: programmed death-ligand 1; TGF-β: transforming growth factor-β; VEGF: vascular endothelial growth factor; YAP: Yes-associated protein; TAZ: transcriptional coactivator with PDZ-binding motif; ADI-PEG20: PEGylated arginine deiminase; Pegargininase: synonym for ADI-PEG20.
Representative trials addressing strategies to overcome resistance to immunotherapy in MPM (2020-2025)
| Representative trials (NCT/year) | Phase/n | Strategy | Key outcomes | Mechanistic rationale | Ref. |
| BEAT-meso (NCT03762018, 2024) | Phase III / n = 400 | Atezolizumab + Bevacizumab + Carboplatin + Pemetrexed vs. Bevacizumab + chemotherapy | ORR: 55% vs. 49% (NS); PFS: 9.2 vs. 7.6 mon (HR = 0.72, P = 0.0021); OS: 20.5 vs. 18.1 mon (HR = 0.84, NS); non-epithelioid subgroup OS HR = 0.51 | VEGF blockade normalizes vasculature, decreases Tregs/M2-TAMs, improves T-cell infiltration | [9,107,108,110] |
| KEYNOTE-483 (NCT02784171, 2023-2024); PrE0505 (NCT02899195, 2022) | Phase III / n = 539; Phase II / n = 55 | Phase III: Pembrolizumab + Platinum/Pemetrexed vs. chemotherapy Phase II: Durvalumab + Platinum/Pemetrexed | Phase III: ORR: 52% vs. 29%; PFS: 7.1 vs. 7.1 mon (NS); OS: 17.3 vs. 16.1 mon Phase II: ORR: 56% (BICR); DCR: 92%; PFS: 6.7 mon; OS: 20.4 mon | Cytotoxic therapy induces ICD and DAMP release to prime DC-T cell axis | [40,89,91,96] |
| ATOMIC-Meso (NCT02709512, 2024) | Phase II/III / n = 249 | Pegargiminase + chemotherapy vs. chemotherapy | PFS: 6.1 vs. 5.6 mon (HR = 0.65); OS: 9.3 vs. 7.7 mon (HR = 0.71) | ASS1-deficient tumors depend on extracellular arginine; deprivation enhances T-cell function | [25,36,80,86] |
| OT-101 + Pembrolizumab (NCT05425576, ongoing) | Phase II / ongoing | TGF-β inhibition + ICI | ORR: not reported PFS: not reported OS: not reported Safety: preliminary data suggest good tolerability | TGF-β drives Treg induction and fibrosis; blockade restores cytotoxic function | [26,27] |
| NCT02414269 (2022, Nat Med); NCT04577326 (ongoing) | Phase I/II / n = 21 + 36 | Cellular immunotherapy (CAR-T) | ORR: 12.5% (2/16 PR); DCR: 68.8%; PFS/OS: not reported | Local delivery of MSLN-targeted CAR-T enhances tumor infiltration; PD-1 inhibition prevents exhaustion | [113,118,126] |
| Gavo-cel (NCT03585764, 2023) | Phase I / n = 39 | TRuC-T therapy | ORR: 21% (BICR) / 26% (investigator); PFS: 5.6 mon; OS: 11.2 mon | TCR-fusion construct enhances immune synapse formation and persistence | [126] |
| ONCOS-102 (NCT02879669, 2020); INFINITE (NCT03710876, 2025) | Phase I / n = 12; Phase III / n = 280 | Oncolytic virus therapy | ONCOS-102: ORR: 30%; DCR: 90% PFS: 9.8 mon; OS: 25.0 vs. 13.5 mon; Safety: well tolerated, no DLT INFINITE: ORR: ~25% (based on early Phase II) DCR: ~87.5% PFS: not reported OS: not reported Safety: manageable (early data) | Viral lysis releases tumor antigens, converts “cold” to “hot” TME | [144,145] |
| DENIM (NCT03610360, 2025); MESOVAX (NCT03546426, 2023) | Phase III / n ≈ 150; Phase I/II / n = 15 | DENIM: DC vaccines MESOVAX: autologous DCs + tumor lysate | DENIM: PFS: 4.1 mon; OS: 11.9 mon MESOVAX: ORR: 56% (BICR); PFS: 6.7 mon; OS: 20.4 mon | DC vaccines enhance antigen presentation and ICI synergy | [40,83,97,99] |
| IK-930 (NCT05228015, ongoing) | Phase I / n ≈ 40 | Oral TEAD inhibitor | ORR/PFS/OS: not reported (early-phase study) | EZH2 or TEAD inhibition restores antigenicity and interferon signaling | [62,63] |
Chemo-immunotherapy to potentiate immunogenic cell death
The combination of conventional cytotoxic chemotherapy with ICIs may seem counterintuitive, given the historical view of chemotherapy as being immunosuppressive. However, a paradigm shift has occurred with the understanding that certain chemotherapeutic agents can induce immunogenic cell death (ICD) platinum-based drugs and pemetrexed, the backbone of MPM chemotherapy, are potent inducers of ICD[88,89]. Representative clinical trials evaluating this strategy are summarized in Table 2. This specialized form of apoptosis is characterized by the exposure and release of damage-associated molecular patterns (DAMPs), including the translocation of calreticulin to the cell surface, the extracellular release of ATP, and the passive secretion of HMGB1[90]. These “danger signals” act as potent adjuvants: they drive the maturation and antigen-presenting function of DCs, enhance the cross-presentation of tumor antigens to naïve T cells, and ultimately amplify the priming and clonal expansion of tumor-specific CD8+ T cells. This process effectively turns chemotherapy into an in situ vaccine, thereby counteracting one of the principal causes of ICI failure: inadequate initial T-cell activation and priming[91-94].
ICIs with combination therapies - updated table (2020-2025)
| Representative trials (NCT/year) | Phase | Patients (n) | Agent/strategy | Key outcomes | Target/pathway | Ref. |
| MAPS2 (NCT02716272, 2019. Updated 2023) | Phase II | 125 | Nivolumab ± Ipilimumab | ORR: 28% (combination); PFS: 3.0 mon; OS: 15.9 mon (combo) vs. 11.9 mon (monotherapy) | PD-1/CTLA-4 dual blockade - T-cell activation | [34] |
| PROMISE-meso (NCT02991482, 2020) | Phase III | 144 | Pembrolizumab vs. single-agent chemo | ORR: 22% vs. 6% (P = 0.004); PFS: 2.5 vs. 3.4 mon (HR = 1.06; P = 0.76); OS: 10.7 vs. 11.7 mon (HR = 1.12; 95%CI 0.74-1.69; P = 0.59) | PD-1 blockade - immune checkpoint inhibition | [105] |
| CheckMate 743 (2021-2023) | Phase III | 605 | Nivolumab + Ipilimumab vs. platinum-pemetrexed | ORR: 40% vs. 44%; median PFS: not consistently reported; 3-year PFS rate: ~14% vs. ~1%; OS: 18.1 vs. 14.1 mon (HR = 0.73); 3-year OS rate: ~23% vs. ~15% | Dual PD-1/CTLA-4 pathway - durable immune memory | [171] |
| KEYNOTE-483 (FDA-approved 2024) | Phase III | Approx.440 | Pembrolizumab + platinum-pemetrexed vs. chemo | ORR: 52% vs. 29%; PFS: 7.1 vs. 7.1 mon; OS: 17.3 vs. 16.1 mon | PD-1 inhibition + cytotoxic synergy | [96] |
| BEAT-meso (NCT03762018, 2025) | Phase III | 400 | Atezolizumab + Bevacizumab + chemo vs. Bevacizumab + chemo | ORR: 55% vs. 49%; PFS: 9.8 vs. 7.6 mon; OS: 25.0 vs. 13.5 mon | PD-L1 blockade + VEGF inhibition - immune-angiogenic synergy | [9,107,108] |
| OT-101 + Pembrolizumab (NCT05425576, Ongoing) | Phase II | - | TGF-β2 antisense (OT-101) + PD-1 inhibitor | ORR: not reported PFS: not reported OS: not reported Safety: preliminary data suggest good tolerability | TGF-β pathway blockade - reversing immune suppression | [26] |
| Avelumab + SBRT (NCT03399552, 2023) | Phase I/II | Small | PD-L1 inhibitor + stereotactic body radiation therapy | ORR/PFS/OS: not reported (safety study) | PD-L1 blockade + radiation-induced antigen release | ClinicalTrials.gov |
| DREAM/DREAM3R (NCT04334759, ongoing) | Phase II/III | - | Durvalumab + platinum-pemetrexed (± maintenance) | ORR/PFS/OS: not reported | PD-L1 blockade + chemo - chemo-immune synergy | [98] |
| NEMO (NCT02863055, ongoing) | Phase III | 116 | Nintedanib + platinum/pemetrexed | ORR/PFS/OS: not reported | Multi-target angiokinase inhibition (VEGFR, PDGFR, FGFR) | [108] |
| ATOMIC-Meso (NCT02709512, 2024) | Phase II/III | 249 | ADI-PEG20 + chemo vs. placebo + chemo | Median OS: 9.3 vs. 7.7 mon (HR = 0.71); Median PFS: 6.2 vs. 5.6 mon | Arginine deprivation - metabolic immunomodulation | [36] |
| IK-930 (NCT05228015, ongoing) | Phase I | - | Oral TEAD inhibitor (Hippo pathway) | ORR/PFS/OS: not reported (early-phase study) | TEAD/YAP-TAZ - tumor suppressor restoration | [62,63] |
| PENINSULA (NCT05412615, 2026 ongoing) | Phase II | Planned n = 25 | Pembrolizumab + Lenvatinib + Platinum/Pemetrexed | ORR/PFS/OS: not reported | PD-1 blockade + VEGFR inhibition + cytotoxic chemotherapy | ClinicalTrials.gov |
| MesoNet | Real-world retrospective | n = 135 | First-line nivolumab + ipilimumab (real-world validation) | OS (overall): 13.1 mon; OS (CM-743 eligible): 15.5 mon; Non-epithelioid OS: 16.7 mon | Dual PD-1/CTLA-4 blockade (real-world confirmation) | [206] |
The randomized, global phase III KEYNOTE-483 (also known as IND227; NCT02784171) trial provides robust clinical validation for this strategy. This pivotal study, which formed the basis for the 2024 FDA approval of the regimen, evaluated pembrolizumab (anti-PD-1) in combination with platinum-pemetrexed chemotherapy vs. chemotherapy alone in 539 patients with previously untreated, unresectable MPM. The combination therapy achieved a modest but clinically meaningful improvement in the primary endpoint of OS, with a median OS of 17.3 months compared to 16.1 months in the chemotherapy-alone arm (HR = 0.79). Consistent with other immunotherapy trials, a more pronounced benefit was observed in the non-epithelioid subgroup. These results validate chemo-immunotherapy as a viable and effective approach to overcoming poor immune priming[95-98].
Further compelling support comes from the single-arm phase II PrE0505 trial (NCT02899195), in which durvalumab (anti-PD-L1) was combined with cisplatin and pemetrexed. This regimen yielded an impressive median OS of 20.4 months - a result that compares favorably with, and in some selected patient subsets may even exceed, outcomes observed with dual ICI regimens. Collectively, the evidence from KEYNOTE-483 and PrE0505 underscores a fundamental principle: that enhancing tumor antigen release and T-cell priming through chemotherapy can effectively mitigate the primary resistance to PD-1/PD-L1 blockade that is observed in a significant number of MPM patients[99-102].
In patients with unresectable MPM who progress after first-line nivolumab plus ipilimumab, effective second-line options remain limited. The phase 2 PEMMELA study (cohort 2) evaluated pembrolizumab plus lenvatinib in this setting and reported an investigator-assessed ORR of 60% (12/20; 95%CI, 36%-81%), with a median PFS of 6.9 months and median OS of 14.1 months. Although grade 3-4 treatment-related adverse events occurred in 70% of patients, these results suggest that immunotherapy combined with targeted therapy may represent a promising strategy to overcome resistance to prior dual immune checkpoint blockade[103-105].
Angiogenesis inhibition to reprogram the TME and restore immunotherapy sensitivity
Pathological angiogenesis is a cornerstone of the immunosuppressive TME in MPM. VEGF, often overexpressed in MPM, does more than simply stimulate the formation of disorganized, leaky, and dysfunctional blood vessels that impede the efficient trafficking of cytotoxic T cells into the tumor core. It also acts as a direct mediator of immune suppression. VEGF signaling inhibits T-cell proliferation and cytotoxic function, impairs the maturation and antigen-presenting capacity of DCs, and actively promotes the recruitment and expansion of Tregs and M2-polarized TAMs. Thus, VEGF blockade represents a dual-pronged therapeutic strategy: it promotes “vascular normalization”, which improves perfusion and T-cell infiltration, while simultaneously dismantling a key immunosuppressive axis within the TME[106-108].
The phase III BEAT-meso trial (NCT03762018) stands as the most definitive clinical evaluation of this approach to date. This randomized study compared the efficacy of atezolizumab (anti-PD-L1) plus bevacizumab (anti-VEGF) and platinum-pemetrexed chemotherapy [the atezolizumab, bevacizumab, and platinum-pemetrexed chemotherapy (ABC) regimen] against bevacizumab plus chemotherapy alone [the bevacizumab plus platinum-pemetrexed chemotherapy (BC) regimen] in 400 previously untreated, unresectable MPM patients. Results, published in Annals of Oncology in 2025, demonstrated a statistically significant and clinically meaningful improvement in the co-primary endpoint of PFS, with a median PFS of 9.2 months in the ABC arm vs. 7.6 months in the BC arm (HR = 0.72). Although the other co-primary endpoint, OS, did not cross the statistical significance boundary (median OS 20.5 vs. 18.1 months; P = 0.14), a critical prespecified subgroup analysis revealed a pronounced OS benefit in patients with non-epithelioid histology (HR = 0.51). This finding reinforces the concept of histology-specific biology. It suggests that the more aggressive, chemoresistant non-epithelioid subtypes, which are often highly angiogenic, may derive exceptional benefit from combined angiogenesis and immune checkpoint inhibition. Mechanistically, bevacizumab-mediated VEGF inhibition has been shown in translational studies to reduce Treg accumulation, inhibit M2 TAM polarization, and facilitate CD8+ T-cell entry into tumor islets, providing a sound biological rationale for this combination. By inhibiting VEGF signaling, this approach not only reverses the immunosuppressive environment driven by aberrant angiogenesis but also restores dysfunctional vascular architecture described in Section MECHANISTIC INSIGHTS INTO THERAPEUTIC ACTION. This dual action provides a mechanistic foundation for its synergistic effect with ICIs, which is particularly pronounced in the highly angiogenic and inherently resistant non-epithelioid subtype. Ongoing studies, such as those evaluating multikinase inhibitors with potent anti-angiogenic properties [e.g., nintedanib in the NEMO trial (NCT02863055, Ongoing)], may help refine our understanding of vascular modulation and reinforce its centrality in reconditioning the TME to support sustained ICI efficacy[109-111].
Metabolic reprogramming as an immune-modulatory therapeutic avenue in MPM
Metabolic competition within the TME constitutes a critical and often overlooked barrier to effective immunotherapy. A hallmark of MPM, particularly the non-epithelioid subtype, is the frequent epigenetic silencing and loss of ASS1, a key enzyme in the de novo synthesis of arginine. This loss renders cancer cells auxotrophic for extracellular arginine, creating a profound metabolic dependency. This dependency has two major implications for ICI resistance. First, it instigates a fierce nutrient competition between rapidly dividing tumor cells and infiltrating T cells, for which arginine is an essential nutrient required for TCR signaling, proliferation, and survival. Second, this competition is exacerbated by elevated arginase-1 activity from MDSCs within the TME, which further depletes local arginine pools, leading to T-cell dysfunction, cell cycle arrest, and functional paralysis[112].
The phase II/III ATOMIC-Meso trial (NCT02709512) was designed to exploit this very vulnerability. The study evaluated the efficacy of pegargiminase [PEGylated Arginine Deiminase (ADI-PEG20)], a microbially derived arginine-degrading enzyme, in combination with first-line platinum–pemetrexed chemotherapy in 249 patients with ASS1-deficient MPM. As reported in JAMA Oncology in 2024, the combination demonstrated statistically significant improvements in both OS (median OS 9.3 vs. 7.7 months; HR = 0.71) and PFS (median PFS 6.2 vs. 5.6 months). While the primary anti-tumor effect of ADI-PEG20 is direct cytotoxicity against ASS1-deficient cancer cells by inducing arginine starvation, an important indirect immunomodulatory effect is now appreciated. By systemically depleting arginine, ADI-PEG20 may reduce the tumor’s nutrient consumption and potentially alter MDSC function, thereby replenishing bioavailable arginine for T-cell activation and attenuating immunosuppressive enzymatic activity in the TME. This may ultimately promote the restoration of T-cell effector functions[36]. This approach directly targets the metabolic vulnerability of ASS1-deficient tumors discussed in Section MECHANISTIC INSIGHTS INTO THERAPEUTIC ACTION, alleviating nutrient competition between cancer cells and tumor-infiltrating lymphocytes (TILs) and providing a mechanistic rationale for combining ADI-PEG20 with ICIs.
The success of ATOMIC-Meso has unequivocally established metabolic immunomodulation as a promising and novel therapeutic avenue in MPM. It suggests that ASS1 status may serve as a potential predictive biomarker for patient stratification, although further prospective validation is warranted.
Cellular immunotherapy to reinvigorate effector function
Adoptive cellular immunotherapy has emerged as one of the most promising strategies to counteract primary and acquired resistance to ICIs in MPM. Unlike ICIs, which rely on the presence of pre-existing functional T cells, adoptive cell therapies directly introduce large numbers of tumor-specific effector lymphocytes capable of overcoming T-cell exclusion, exhaustion, and suppressive stromal barriers. The pleural cavity offers a unique anatomic advantage for regional delivery, allowing high local concentrations of engineered cells while minimizing systemic toxicities typically associated with CAR-T therapies[36,113].
An overview of key efforts in cellular immunotherapy for MPM is summarized in Tables 3 and 4 and Figure 3.
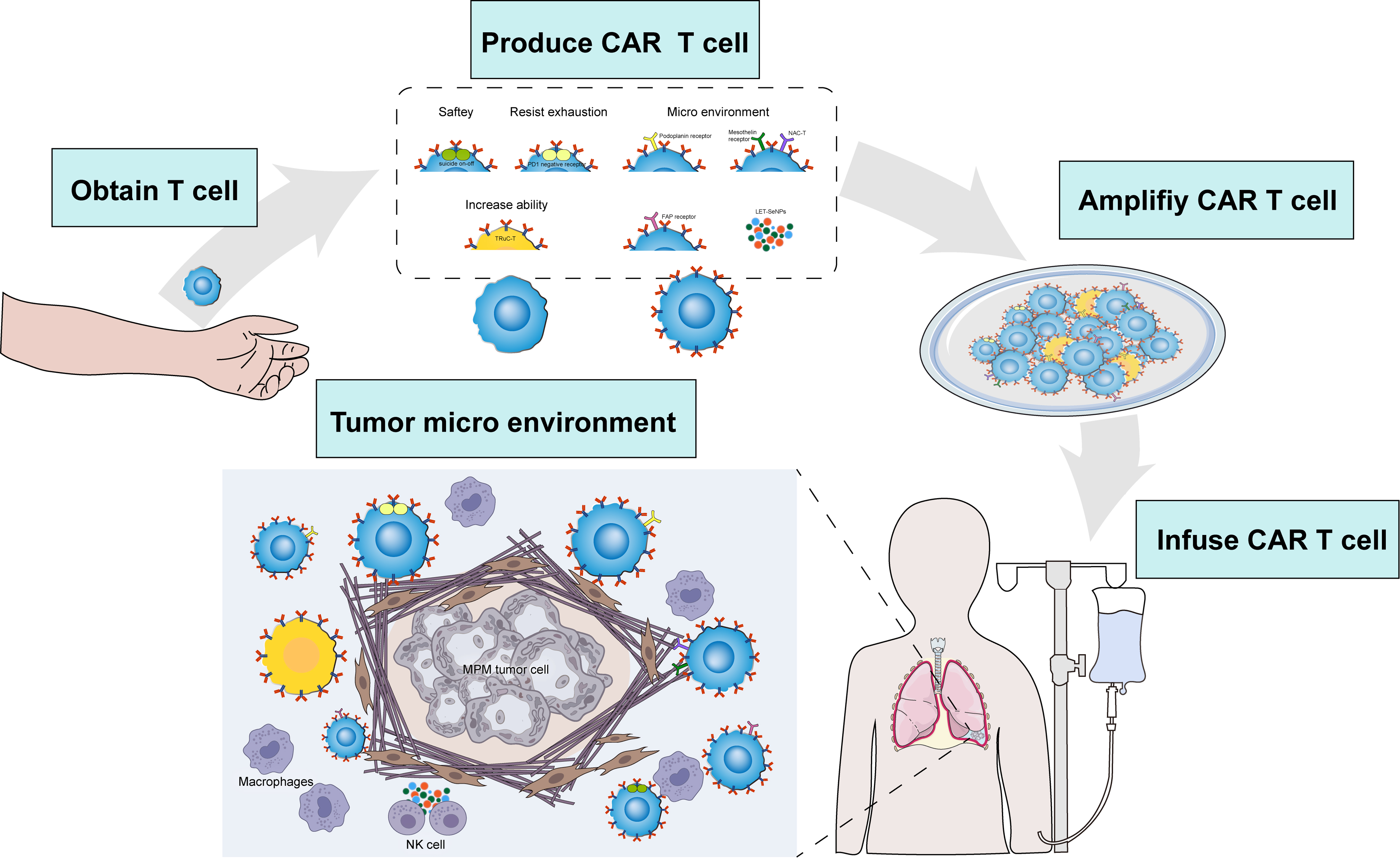
Figure 3. Cellular immunotherapy for MPM. MPM: Malignant pleural mesothelioma; CAR-T: chimeric antigen receptor T cells; PD1: programmed cell death protein 1; NAC-T: nanobody-armored CAR-T cells; TRuC-T: T-cell receptor fusion construct; FAP: fibroblast activation protein; LET-SeNPs: lentinan-functionalized selenium nanoparticles; NK: natural killer.
CAR-T (regional with engineering strategies)
| Representative trials (NCT/year) | Phase | Patients (n) | Agent/strategy | Key outcomes | Target/pathway | Ref. |
| NCT02414269 (MSKCC, 2021 ASCO; 2022 Nat Med) | Phase I/II | n = 21 (19 MPM) | iCasp9 M28z CAR-T (anti-MSLN) + Pembrolizumab | ORR: 12.5% (2/16 PR); DCR: 68.8%; PFS/OS: not reported | MSLN CAR-T + PD-1 blockade | [113] |
| NCT03054298 (M5 CAR, 2022-2023 ASCO/SITC) | Phase I | Planned n = 18 | Fully humanized MSLN CAR-T | ORR: not reported; PFS: not reported; OS: not reported | MSLN (humanized scFv, low immunogenicity) | [115-117] |
| NCT04577326 (ATA2271, 2023 ASCO) | Phase I | Planned n = 36 (ongoing) | PD-1–blocked MSLN CAR-T (intrapleural) | ORR: not reported; PFS: not reported; OS: 23.9 months (reported at ASCO 2023); 1-year OS: 83% | MSLN + PD-1–resistant CAR-T | [118] ClinicalTrials.gov |
| NCT03585764 (gavo-cel/TRuC-T, 2022 ASCO; 2023 Clin Cancer Res) | Phase I | n = 39 | TRuC-T therapy | ORR: 21% (BICR) / 26% (investigator); PFS: 5.6 mon; OS: 11.2 mon | MSLN-targeted TRuC platform | [126] |
| NCT01722149 (FAP CAR-T, 2014 Sci Transl Med) | Phase I | n = 3 | CAR-T targeting FAP | ORR: not reported; PFS: not reported; OS: not reported; No CRS observed | FAP (stromal target) | [119,120] |
| NCT03618381 (PDPN CAR-T, expected 2025) | Phase I | Planned n = 18 (ongoing) | CAR-T targeting PDPN | ORR: not reported; PFS: not reported; OS: not reported | PDPN | [121] ClinicalTrials.gov |
| FIH study (2023) | Phase I | n = 11 | NAC-T (MSLN CAR-T armored with anti-PD-1 nanobody) | ORR: 63.6%; DCR: 100%; OS: 25.6 months; PFS: not reported; No DLT observed | MSLN/local PD-1 blockade | [127] |
| Published preclinical study (2025) | Preclinical | - | LET-SeNPs | Restored NK cytotoxicity, upregulated NKG2D expression, and inhibited invasion in spheroid and organoid models | TrxR1–pSTAT3/NKG2D axis | [128] |
Dendritic-cell vaccines with oncolytic/viral-based cytokine delivery
| Representative trials (NCT/year) | Phase | Patients (n) | Agent/strategy | Key outcomes | Target/pathway | Ref. |
| Autologous DC vaccines (Multiple Trials, 2018-2021) | Phase II (various) | > 100 (across studies) | Autologous DCs loaded with tumor lysate or defined antigens ± ICI | mOS: 19-27 months; 2-year OS ≈ 50%-55%; durable immune responses; mainly grade 1-2 AEs | Antigen presentation → T-cell activation | [136-138] |
| NCT03175172 (CRS-207 + Pembrolizumab, 2020 Cancers) | Phase II | Small cohort (not disclosed) | Live-attenuated Listeria-based MSLN vaccine + PD-1 inhibitor | No significant clinical activity; trial terminated | MSLN vaccine + PD-1 blockade | ClinicalTrials.gov |
| NCT00327652/NCT00358945 (CRS-207 + Chemo, 2007-2012) | Phase I/Ib | n = 18 + 35 | CRS-207 combined with pemetrexed/platinum chemo | DCR: 89%; no OS improvement in later expansion | Vaccine-induced immune priming + chemo | ClinicalTrials.gov |
| NCT02879669 (ONCOS-102, 2019 J Immunother Cancer) | Phase I | n = 12 | GM-CSF–expressing oncolytic adenovirus + chemo | ORR: 30%; DCR: 90%; mPFS: 9.8 months; mOS: 25.0 months (vs. 13.5 control) | Oncolysis + T-cell infiltration enhancement | [143-145] |
| NCT01119664 (Ad-IFN, 2014-2018 series) | Phase I | n = 21 | Intrapleural adenoviral IFN-β gene therapy | DCR: 67%; mOS: 21.5 months; manageable safety | Local IFN signaling → immune activation | [139-142] |
| NCT01721018 (HSV1716, 2018 Gene Therapy) | Pilot | n = 21 | Intrapleural oncolytic HSV1716 | ORR: 0%; 50% stable disease; immune activation observed | Oncolytic HSV → local immune stimulation | [143,147-149] |
| NCT03546426 (MESOVAX)/NCT05765084 (Immuno-MESODEC, 2022-2023 conference) | Phase I/II | MESOVAX n = 15; Immuno-MESODEC planned n = 36 | Autologous DC vaccine + Pembrolizumab WT1mRNA-electroporated DC + Atezolizumab + chemo | NCT03546426: ORR: ~56% (BICR); mPFS: 6.7 months; mOS: 20.4 months NCT05765084: Ongoing; immune activation signals reported; survival data pending | DC activation + PD-1 blockade WT1 antigen presentation + PD-L1 inhibition | [99,135] ClinicalTrials.gov |
Across cellular immunotherapy strategies, mesothelin (MSLN)-directed platforms remain the dominant target. Early-phase CAR-T studies demonstrate modest ORR (12%-26%) with limited durability data, whereas armored or PD-1–resistant constructs (e.g., NAC-T) show substantially higher response rates in small cohorts (ORR 63.6%; mOS 25.6 months), suggesting that overcoming intratumoral immune suppression is critical. TRuC-T therapy demonstrates measurable activity with defined median PFS (5.6 months) and OS (11.2 months), providing the most mature survival data among MSLN-targeted cellular therapies to date.
The most mature evidence comes from MSLN-directed CAR-T cells. The landmark phase I/II trial conducted at Memorial Sloan Kettering Cancer Center (MSKCC) (NCT02414269) evaluated intrapleural infusion of inducible caspase 9 (iCasp9)-mesothelin-targeting CAR with CD28 and CD3ζ signaling domains (M28z) CAR-T cells - incorporating a CD28 co-stimulatory domain and a safety “suicide switch” - followed by systemic pembrolizumab. Among 21 heavily pretreated patients (including 19 with MPM), this regimen produced unprecedented outcomes for relapsed disease, with a median OS of 23.9 months, two complete metabolic responses, and durable CAR-T persistence beyond 100 days in nearly 40% of patients. Correlative studies demonstrated increased TILs and reduced markers of exhaustion following PD-1 blockade, providing mechanistic support for sequential combination ICI therapy and establishing a blueprint for CAR-T–ICI integration in MPM[109,114,115]. By supplying large numbers of tumor-specific effector cells directly into the pleural cavity, cellular immunotherapies circumvent the T-cell exclusion and suppressive cellular barriers (Tregs, MDSCs) detailed in Section MECHANISTIC INSIGHTS INTO THERAPEUTIC ACTION, reinforcing the rationale for combining CAR-T cells with checkpoint inhibitors to sustain anti-tumor immunity. Next-generation MSLN-directed platforms aim to improve T-cell persistence, reduce immunogenicity, and overcome the profoundly suppressive pleural TME. The phase I study of human CAR modified T cells in patients with mesothelin expressing cancers (NCT03054298), employing a fully human scFv to minimize anti-murine immune responses, has demonstrated feasible expansion and sustained persistence in early-phase studies, with a favorable safety profile. ATA2271 (NCT04577326) represents a further evolution of this concept: these CAR-T cells incorporate an undetermined signaling backbone and an intrinsic PD-1 dominant-negative receptor to resist exhaustion. Early findings show robust expansion and persistence in low-dose cohorts, although a fatal serious adverse event (SAE) in the high-dose cohort underscores the importance of careful dose optimization in future development[116-118].
Beyond MSLN, several trials have explored alternative antigenic targets relevant to the unique stromal and mesothelial biology of MPM. The FAP-CAR-T trial (NCT01722149) targeted fibroblast activation protein (FAP), a key mediator of the dense fibrotic matrix characteristic of MPM. Despite the small sample size (n = 3), the study demonstrated intratumoral CAR-T expansion and survival beyond 18 months in two patients, highlighting the potential of stromal-directed strategies to remodel the TME and reverse immune exclusion. Similarly, podoplanin (PDPN)-CAR-T therapy (NCT03618381), targeting PDP - another clinically relevant mesothelial antigen - has entered phase I evaluation, with ongoing analyses focused on trafficking, biodistribution, and safety[115,119-122].
Complementing traditional CAR-T constructs, the TCR fusion construct T (TRuC-T) cell platform represents an innovative approach that integrates CAR specificity into the native TCR complex. In the phase I trial of gavo-cel (NCT03585764), 39 patients with advanced MPM received TRuC-T cells targeting MSLN, achieving an ORR of 21%, median PFS of 5.6 months, and median OS of 13.7 months. These findings highlight the enhanced signaling potency and physiologic activation afforded by TRuC designs, offering a potentially superior alternative to conventional second-generation CAR-T architectures. To further counteract CAR-T exhaustion within the immunosuppressive MPM microenvironment, MSLN-targeted CAR-T cells armored with an anti-PD-1 nanobody [nanobody-armored CAR-T cells (NAC-T)] were developed. In a first-in-human study involving 11 patients with refractory malignant mesothelioma, intravenous infusion of 5 × 106-20 × 106 cells/kg NAC-T achieved an ORR of 63.6%, DCR of 100%, median PFS of 5.0 months, and median OS of 25.6 months, without dose-limiting toxicity. Single-cell and TCR sequencing demonstrated clonal expansion of tumor-reactive T-cell subsets, underscoring the therapeutic potential of locally enhanced checkpoint blockade within engineered cellular platforms. Beyond adaptive T-cell engineering, nanotechnology-based approaches are emerging to reprogram innate immunity and metabolic vulnerabilities within the MPM microenvironment. Recent evidence indicates that MPM-associated pleural effusions exhibit selenium deficiency and reduced selenoprotein expression [e.g., selenoprotein P (SEPP1)], correlating with impaired natural killer cell (NK-cell) function. To address this redox imbalance, lentinan-functionalized selenium nanoparticles (LET-SeNPs) were developed as immunomodulatory agents capable of restoring bioavailable selenium locally. Mechanistically, LET-SeNPs activate the Thioredoxin reductase 1–phosphorylated signal transducer and activator of transcription 3 (TrxR1–pSTAT3) axis, upregulate natural killer group 2, member D (NKG2D) expression, and enhance NKG2D–NKG2D ligands (NKG2DL)–mediated cytotoxicity. Preclinical spheroid and organoid models demonstrated improved NK-cell infiltration, reduced invasive capacity, and decreased matrix metalloproteinase-2 (MMP-2) expression. Together, these findings suggest that metabolic trace-element reprogramming may synergize with engineered T-cell strategies to reinforce multi-layered immune control in MPM[123-129].
Collectively, the expanding portfolio of adoptive cellular therapies in MPM reflects a multipronged attempt to overcome the immunologic bottlenecks that limit ICI efficacy: supplying effector cells to overcome low T-cell infiltration, engineering exhaustion-resistant phenotypes, targeting both tumor and stromal antigens to dismantle physical and biochemical barriers, and integrating CAR-T with ICIs to sustain antitumor immunity. As translational advances continue to refine antigen selection, delivery strategies, and resistance-proof CAR architectures, cellular immunotherapy is poised to play a central role in next-generation therapeutic combinations designed to surmount the deeply entrenched immune resistance landscape of MPM[130-132].
A fundamental contributor to primary resistance to ICIs in MPM is the failure of adequate antigen presentation, which results in insufficient priming and expansion of tumor-reactive T cells. DC vaccination seeks to directly correct this deficit by ex vivo generation of professional antigen-presenting cells. This approach typically involves isolating autologous monocyte-derived DC precursors, maturing them with defined adjuvants, and pulsing them with tumor-associated antigens - either whole tumor lysate or specific antigens such as Wilms tumor 1 (WT1) - before reinfusion. Upon administration, these antigen-loaded DCs traffic to secondary lymphoid organs, where they efficiently prime naïve CD4+ and CD8+ T cells, generating a broad, polyclonal, and durable antitumor immune response capable of synergizing with ICIs[133-136].
The ongoing phase II/III dendritic cell immunotherapy for mesothelioma (DENIM) trial (NCT03610360) is one of the key ongoing clinical studies evaluating this strategy in MPM. As a randomized, placebo-controlled study testing tumor-lysate–pulsed DC vaccination as maintenance therapy following first-line chemotherapy, DENIM builds on earlier phase I/II signals in which autologous DC vaccination produced durable antigen-specific immunity and median OS reaching up to 27 months in selected cohorts. Parallel early-phase studies now explore rational DC–ICI combinations. The pembrolizumab plus autologous dendritic cell vaccine in patients with PD-L1 negative advanced mesothelioma who have failed prior therapies (MESOVAX) phase I trial (NCT03546426) evaluates autologous DC vaccination administered concurrently with pembrolizumab in previously treated MPM, aiming to enhance T-cell priming while preventing subsequent PD-1–mediated exhaustion. Initial data indicate that the regimen is feasible, immunogenic, and capable of inducing disease stabilization in a subset of patients. In frontline disease, the Immuno-mesothelioma dendritic cell (MESODEC) trial (NCT05765084) is testing a triplet strategy combining WT1-targeted DC vaccination with atezolizumab and standard chemotherapy, leveraging chemotherapy-induced ICD, WT1-specific T-cell activation, and PD-L1 blockade to maximize priming and effector maintenance. Collectively, these trials reflect a mechanistically coherent development path in which DC vaccines augment antigen presentation while ICIs sustain effector functionality within the chronically immunosuppressive MPM microenvironment, supporting their integration into multimodal immunotherapeutic strategies[26,40,135,137-139].
Oncolytic viruses to convert “cold” to “hot” tumors
A major obstacle to effective immunotherapy in MPM is the predominance of immunologically “cold” tumors, characterized by limited T-cell infiltration and weak antigenicity. Oncolytic viruses offer a compelling strategy to overcome this barrier by acting as in situ vaccines: they selectively infect tumor cells, induce ICD, and release a diverse array of neoantigens, DAMPs, and viral PAMPs that ignite innate and adaptive immune responses. The resulting interferon beta (IFN-β) enhances dendritic-cell activation, promotes chemokine-driven T-cell recruitment, and primes de novo antitumor immunity - thereby establishing a more permissive microenvironment for subsequent ICI therapy[133,140-143].
The GM-CSF–expressing adenovirus a randomised open-label phase I/II study adding ONCOS-102 to pemetrexed/cisplatin in patients with unresectable malignant pleural mesothelioma (ONCOS-102) (NCT02879669) has demonstrated these immunostimulatory properties in early-phase clinical studies. In combination with chemotherapy, ONCOS-102 markedly increased intratumoral CD8+ T-cell infiltration and upregulated immune-activation gene signatures, with the most pronounced immunologic changes correlating with clinical benefit. Notably, exploratory follow-up analyses revealed upregulation of PD-L1 after ONCOS-102, providing a mechanistic rationale for combining or sequencing with PD-1 inhibitors. Early clinical experiences using ONCOS-102 followed by pembrolizumab reported sustained T-cell expansion and durable tumor control, supporting a synergistic “prime-and-boost” paradigm wherein oncolytic viruses initiate immune activation and ICIs maintain effector function[144-146].
Beyond adenoviral platforms, the oncolytic herpesvirus HSV1716 (Oncolytic Herpes Simplex Virus type 1 strain 1716; NCT01721018) has shown favorable safety and an ability to stimulate localized immune activation in MPM. This lays the groundwork for future strategies combining herpes simplex virus (HSV)-based therapy with PD-1/PD-L1 blockade. Preclinical studies further reinforce these concepts: the cytokine-armed adenovirus Ad5/3-E2F-D24-hTNFα-IRES-hIL-2 (TILT-123), encoding TNF-α and IL-2, synergized with anti–PD-1 therapy in mouse models, inducing systemic “abscopal-like” tumor regression and durable immunologic memory. These findings highlight the broader potential of oncolytic immunotherapy to reprogram the mesothelioma microenvironment, thereby converting inherently cold tumors into ICI-responsive lesions[147-151].
Emerging biological pathways and targetable regulators of resistance
Beyond the more established mechanisms discussed above, several foundational biological pathways are increasingly recognized as critical contributors to ICI resistance in MPM, presenting a rich landscape for novel therapeutic intervention[27].
Genomic Determinants: Specific somatic alterations shape the immune contexture: CDKN2A deletion, one of the most common genomic events in MPM, has been associated with intrinsic resistance to PD-1 blockade, potentially through the suppression of interferon signaling pathways that are crucial for antigen presentation. In contrast, BAP1 loss, another frequent occurrence, is paradoxically linked to a more “inflamed” TME characterized by increased immune infiltration and may predict enhanced ICI sensitivity in certain contexts, highlighting the complex interplay between driver mutations and anti-tumor immunity[152].
Metabolic Immunosuppression: Multiple metabolic pathways beyond arginine contribute to the immunosuppressive TME. These include lactate accumulation from aerobic glycolysis, which acidifies the TME and directly impairs T-cell effector function and cytokine production; indoleamine 2,3-dioxygenase 1 (IDO1)-mediated tryptophan depletion, which activates immunosuppressive pathways in T cells; and the activation of the potent adenosine pathway, where the ectoenzymes CD39 and CD73 generate extracellular adenosine that suppresses T cells and NK cells via the Adenosine A2A receptor[27,153-155].
Wingless-related integration site (Wnt)/β-catenin Signaling: The aberrant activation of this developmental pathway is a recognized non-mutational driver of T-cell exclusion in many cancers, including a subset of MPM. It prevents the recruitment of CD103+ DCs and effector T cells into the tumor, creating an “immune-desert” phenotype[156-159].
TGF-β Signaling: TGF-β is a master regulator of the fibrotic, immune-suppressed TME in MPM. It is a potent inducer of Treg differentiation and a key activator of CAFs, which drive the deposition of a collagen-rich extracellular matrix that physically excludes T cells. This pathway is now being actively targeted in the clinic, for example, with the TGF-β inhibitor OT-101 in combination with pembrolizumab (NCT05425576)[160-163]. Inhibition of TGF-β signaling reverses the fibrotic stromal barrier and Treg accumulation highlighted in Section MECHANISTIC INSIGHTS INTO THERAPEUTIC ACTION, representing a rational strategy to remodel the TME and enhance ICI efficacy.
Multiple immunotherapeutic platforms are currently being explored in MPM. To date, dual immune checkpoint blockade has demonstrated a statistically significant OS improvement in a phase III setting. This level of evidence offers a comparative context for interpreting the clinical development of other investigational approaches. Metabolic interventions, such as arginine depletion in ASS1-deficient tumors, illustrate the promise of biomarker-driven stratification but have yet to produce consistent survival gains at the population level. Their efficacy appears context-dependent and vulnerable to adaptive metabolic rewiring. Similarly, MSLN-targeted armored CAR-T constructs generate striking early response rates and encouraging survival signals; however, these findings derive from small, non-randomized cohorts and require validation in adequately powered trials.
Regional approaches, including tumor treating fields (TTFields), produce survival outcomes comparable to historical chemotherapy standards but lack direct comparison with immune checkpoint blockade. At the preclinical level, immunometabolic reprogramming strategies, including nanoparticle-mediated restoration of natural killer cell cytotoxicity, underscore the mechanistic sophistication of the field, yet remain translationally immature.
Nevertheless, several significant limitations must be taken into account. The majority of combination strategies for MPM are still in early-phase clinical development, often relying on small, non-randomized cohorts without biomarker-driven patient stratification. Consequently, the reported response signals may be more reflective of selection bias rather than genuine mechanistic synergy. In addition, overlapping toxicities, high costs, and logistical complexities - especially for cellular and regionally delivered therapies - present formidable translational hurdles. Currently, many combination approaches are empirically constructed based on biological plausibility rather than prospectively validated resistance mechanisms. Future progress will hinge on rigorous randomized validation, adaptive trial designs, and robust biomarker integration to differentiate rational synergy from mere additive experimentation. Moving forward, real progress will hinge not just on developing new biological concepts, but on integrating robust biomarkers, designing smarter combinations, and confirming durable survival benefits through rigorous randomized trials - rather than relying on early-phase signals[129,164,165].
BIOMARKERS AND PRECISION IMMUNO-ONCOLOGY IN MPM
From empiricism to precision
MPM has historically been managed with empiric therapeutic strategies, resulting in consistently poor outcomes across its diverse histologic and molecular subtypes. The introduction of ICIs, specifically anti–PD-1 and anti–CTLA-4 antibodies, has marked the beginning of a new therapeutic epoch. Nonetheless, despite transformative benefits in certain patient subgroups, overall response rates remain modest - approximately 23%-25% for monotherapy and 35%-40% for dual immunotherapy combinations, as demonstrated in the CheckMate 743 and MAPS2 trials[166].
This limited and heterogeneous clinical efficacy underscores the pressing need for predictive and prognostic biomarkers to optimize patient selection, treatment sequencing, and rational combination therapy design, as summarized in the integrative framework in Figure 4. Unlike melanoma or non-small cell lung cancer (NSCLC), MPM is characterized by a low TMB, a restricted neoantigen landscape, and a highly complex TME dominated by immune exclusion and stromal components - all of which pose significant challenges to biomarker development[167-169].
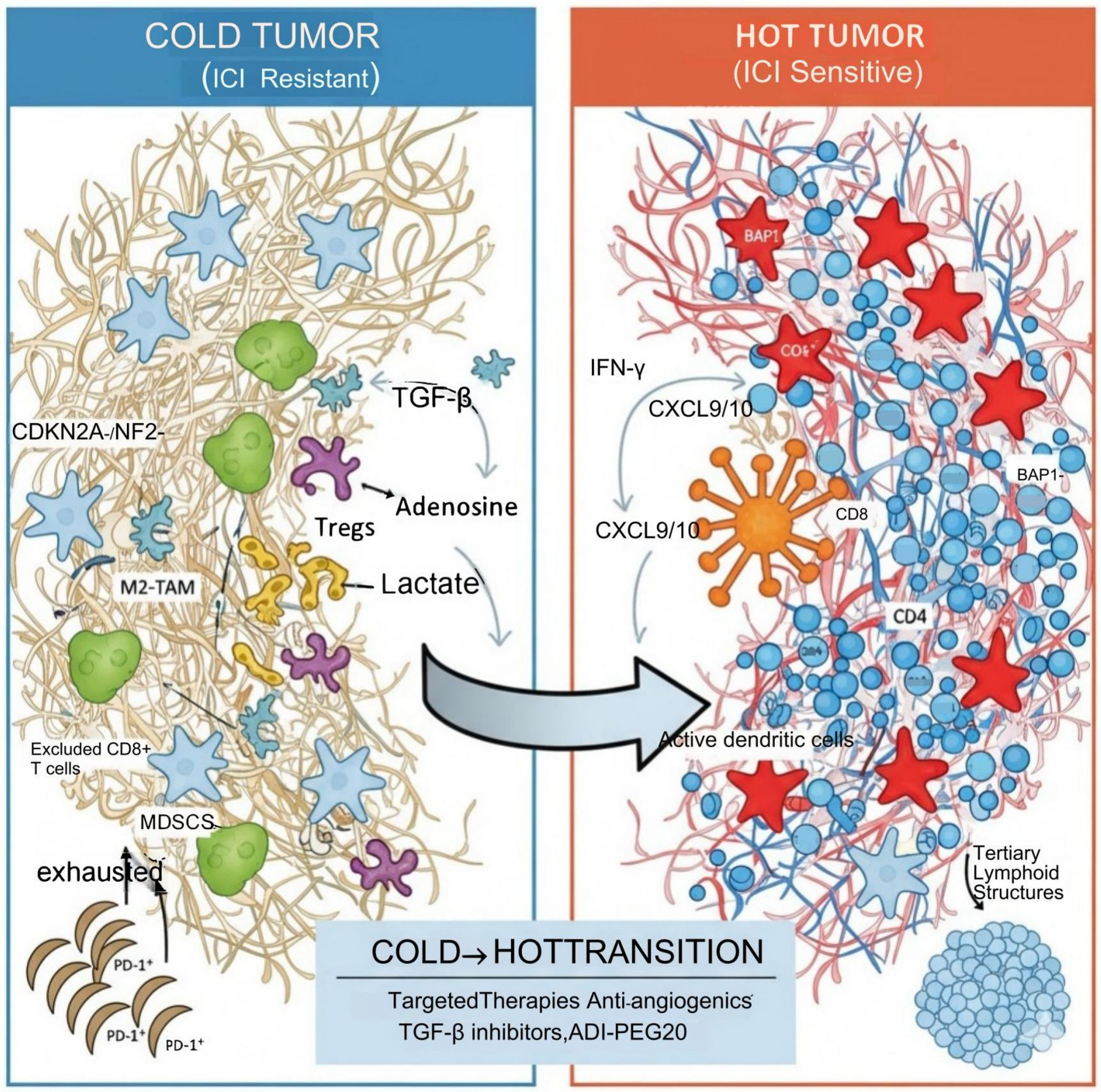
Figure 4. MPM TME: from “cold” to “hot”. MPM: Malignant pleural mesothelioma; TME: tumor microenvironment; ICI: immune checkpoint inhibitor; CDKN2A: cyclin-dependent kinase inhibitor 2A; NF2: neurofibromin 2; TGF-β: transforming growth factor beta; Tregs: regulatory T cells; M2-TAM: M2-type tumor-associated macrophage; CD8+ T cells: cytotoxic T lymphocytes; MDSCs: myeloid-derived suppressor cells; PD-1: programmed cell death protein 1; IFN-γ: interferon γ; CXCL9/10: chemokine (C-X-C motif) ligand 9/10; BAP1: BRCA1-associated protein 1; ADI-PEG20: PEGylated arginine deiminase.
Established biomarkers: PD-L1, TMB, and histologic context
PD-L1 expression, although widely assessed, demonstrates inconsistent predictive utility in MPM. In the CheckMate 743 trial, PD-L1 expression ≥ 1% was correlated with improved OS in the nivolumab–ipilimumab group (median OS 18.0 vs. 13.3 months; HR = 0.69); however, clinical benefit was also observed in PD-L1–negative patients, indicating its limited discriminatory power. Furthermore, inter-assay variability (e.g., between the PD-L1 IHC 22C3 and PD-L1 IHC SP263 antibody clones) and significant intratumoral heterogeneity further compromise the reliability of PD-L1 as a standalone biomarker[170,171].
Similarly, TMB in MPM is generally low, typically below 2 mutations/Mb, which aligns with its non-mutagenic, asbestos-driven etiology. As a result, TMB alone is inadequate for predicting ICI responsiveness. However, specific mutational signatures - such as those associated with defective DNA damage repair (DDR) and alterations in chromatin-remodeling genes [e.g., BAP1, NF2, SET domain containing 2 (SETD2)] - may hold greater immunologic relevance[172-174].
Histologic subtype remains the most clinically actionable biomarker to date. Non-epithelioid MPM, despite its historically unfavorable prognosis, derives disproportionate benefit from dual ICI regimens (e.g., in CheckMate 743: OS 18.1 vs. 8.8 months; HR = 0.46), likely attributable to higher baseline immune infiltration and inflamed transcriptional profiles[175,176].
Genomic and transcriptomic biomarkers
Beyond conventional markers, comprehensive genomic profiling has uncovered a spectrum of mutations and transcriptional phenotypes that critically influence immune responsiveness.
BAP1 loss, occurring in 50%-60% of MPM cases, promotes an “inflamed” tumor phenotype, characterized by enhanced interferon γ (IFN-γ) signaling, elevated chemokine (C-X-C motif) ligand 9/10 (CXCL9/CXCL10) expression, and increased infiltration of cytotoxic lymphocytes. Retrospective analyses indicate improved response rates to ICIs in BAP1-deficient tumors (e.g., as seen in NCT01773655 and correlative studies from CheckMate 743)[177].
Conversely, CDKN2A deletion - found in approximately 70% of cases - is associated with resistance to PD-1 blockade, likely mediated through impaired interferon signaling and downregulation of MHC-I expression. Similarly, dysregulation of the NF2/Hippo pathway contributes to immune evasion via YAP-mediated induction of PD-L1 expression and suppression of pro-inflammatory cytokines[178-181].
Transcriptomic classifications have further refined the distinction between “hot” and “cold” immune phenotypes. Tumors exhibiting enriched immune gene signatures - including elevated expression of chemokine (C-X-C motif) ligand 13 (CXCL13), granzyme (GZMB), and human leukocyte antigen (HLA) genes - correlate with improved OS and greater ICI responsiveness[152,182].
Epigenetic and metabolic biomarkers
Emerging evidence underscores the role of epigenetic dysregulation as both an oncogenic driver and a key determinant of immunogenicity. BAP1 loss, SETD2 mutations, and enhancer of zeste homolog 2 (EZH2) overexpression collectively establish a repressive chromatin state that suppresses antigen presentation. Pharmacologic intervention with HDAC or EZH2 inhibitors (e.g., tazemetostat, NCT02860286) can restore MHC-I expression and promote immune infiltration, highlighting epigenetic modulation as a promising biomarker-guided therapeutic avenue[183-186].
On the metabolic front, arginine metabolism has arisen as a critical regulator of immunotherapy response. ASS1-deficient tumors display arginine auxotrophy, initiating a metabolic competition with infiltrating T cells for essential nutrients. Clinical trials such as ATOMIC-Meso (NCT02709512) have demonstrated that systemic arginine depletion via ADI-PEG20 not only targets tumor metabolism but also reprograms the TME to favor anti-tumor immunity. Other metabolic pathways - including lactate accumulation, IDO activation, and adenosine signaling - have also been implicated in immune suppression and may serve as composite metabolic biomarkers[187-191].
Circulating biomarkers and liquid biopsy approaches
Liquid biopsy technologies enable noninvasive, longitudinal biomarker assessment through multiple complementary approaches. Soluble PD-L1 levels in plasma correlate with tumor PD-L1 expression and poorer prognosis in retrospective analyses. Meanwhile, circulating tumor DNA and exosomal RNA facilitate dynamic monitoring of mutational burden and immune-related gene expression patterns, including interferon response signatures. Additionally, peripheral immune profiling - particularly an elevated baseline CD8+/Treg ratio and expanded TCR clonality - shows association with improved survival in patients undergoing ICI therapy, as demonstrated in the NCT04577326 translational substudy. These integrated liquid biopsy modalities are currently undergoing prospective validation in ongoing trials such as MESOTHRIVE (NCT04990777) and IMMUNO-MESO (NCT05765084)[192-195].
Integrative and artificial intelligence-driven biomarker discovery
Artificial intelligence (AI)–driven digital pathology is emerging as a critical enabler of precision stratification. Deep learning models applied to whole-slide images can achieve expert-level tumor detection and grading while inferring molecular features such as microsatellite instability (MSI) status, PD-L1 expression, and oncogenic mutations directly from routine histology. Integrative AI frameworks combining histomorphology, multi-omics, radiomics, and clinical data may facilitate dynamic risk prediction and personalized treatment allocation.
However, translation into routine practice will require prospective validation, standardized data harmonization, interpretability improvements, and regulatory alignment. Ultimately, the convergence of engineered immunotherapies, nanomedicine-based microenvironment modulation, and AI-guided patient stratification may redefine the therapeutic landscape of MPM, shifting from empiric management toward adaptive, biology-driven precision oncology[196-199].
Clinical translation and future directions
Despite notable advances, the translation of biomarkers into routine clinical practice confronts several obstacles: inter-assay variability, limited tissue availability, and small, histologically heterogeneous trial cohorts. Looking ahead, adaptive biomarker-guided trials - exemplified by the platform design of PRISM-Meso (NCT05022634) - aim to stratify patients in real time based on immune-genomic characteristics. Moreover, AI-driven predictive models that integrate radiomic features with molecular signatures (i.e., radiogenomics) offer promising avenues for noninvasive biomarker prediction.
Ultimately, the convergence of molecular precision, immunologic insight, and computational analytics is poised to redefine MPM management, steering the field away from empirical immunotherapy toward a future of truly personalized immuno-oncology[200,201].
CONCLUSION
MPM remains a biologically aggressive and therapeutically refractory malignancy. Although immune checkpoint blockade - most notably nivolumab plus ipilimumab, as established in CheckMate 743 (median OS 18.1 months) - has reshaped first-line management, durable clinical benefit remains limited to a subset of patients, reflecting the layered, adaptive, and spatially organized mechanisms of immune resistance within the pleural TME.
A mechanism-guided therapeutic framework is therefore critical. T-cell exhaustion, NK-cell dysfunction, metabolic liabilities such as arginine auxotrophy, stromal fibrosis, aberrant angiogenesis, and dysregulated pathways including TGF-β and Hippo signaling collectively generate an immunosuppressive niche that constrains therapeutic efficacy. These vulnerabilities have catalyzed the development of multidimensional strategies, including exhaustion-resistant and armored CAR-T/TRuC-T platforms, stromal- and antigen-targeted cellular therapies, metabolic deprivation approaches (e.g., ADI-PEG20 in ASS1-deficient tumors), epigenetic modulation to restore antigen presentation, vascular normalization strategies, and biomaterial-enabled locoregional delivery systems. Emerging nanotechnology-based interventions - such as selenium nanoparticle–mediated redox reprogramming to enhance NK-cell cytotoxicity, recently investigated in Chinese translational studies - further exemplify how metabolic and innate immune modulation may synergize with engineered cellular platforms. Regional modalities including TTFields have also demonstrated encouraging survival benefits without added systemic toxicity in phase II evaluation, suggesting that mitotic stress–induced tumor immunogenicity may complement immunotherapeutic strategies.
Concurrently, advances in AI–assisted histopathology, spatial transcriptomics, multi-omics integration, and liquid biopsy technologies are refining biomarker discovery and patient stratification. The convergence of spatial biology and computational modeling holds promise for aligning specific resistance mechanisms with rationally selected combinatorial regimens in a precision oncology framework.
Looking forward, sustained progress will depend on biomarker-enriched and adaptively designed clinical trials, deeper integration of immunology, bioengineering, and computational sciences, and broader geographic representation of enrolled populations. While pivotal trials have been conducted predominantly in Western cohorts, expanding high-quality clinical and translational studies in Asian populations - including cellular and nanotechnology-based immunotherapies - will be essential to validate biomarker-driven strategies across diverse genetic and environmental backgrounds. Linking mechanism to target and to therapeutic strategy provides a coherent translational roadmap and offers the most credible path toward achieving durable immune control in this surface-spreading malignancy[86,202-206].
DECLARATIONS
Acknowledgments
We appreciate and acknowledge the support from mentors for their guidance.
Authors’ contributions
Conceived the contents and structure: Zhang Y, Zhao J, Tan X, Ju S, Zou W, Chen C, Li C, Xu Y, Peng Y
Wrote the original manuscript: Xia W
Guided and improved the manuscript: Li S
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work received the following support. The work of Shengqiao Li was partially supported by the Guangdong Bureau of Traditional Chinese Medicine Research Project (20211079) and a Basic Science project from the Zhuhai Bureau of Technology and Innovation (2220004000322).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Dhalluin X, Scherpereel A. Treatment of malignant pleural mesothelioma: current status and future directions. Monaldi Arch Chest Dis. 2010;73:79-85.
2. Shamseddin M, Obacz J, Garnett MJ, Rintoul RC, Francies HE, Marciniak SJ. Use of preclinical models for malignant pleural mesothelioma. Thorax. 2021;76:1154-62.
3. Chen S, Zhao C, Liu R, Jiao W. A bibliometric analysis of malignant pleural mesothelioma from 2010 to 2023. J Thorac Dis. 2025;17:2014-27.
4. Amin W, Linkov F, Landsittel DP, et al. Factors influencing malignant mesothelioma survival: a retrospective review of the National Mesothelioma Virtual Bank cohort. F1000Res. 2018;7:1184.
5. Kondola S, Manners D, Nowak AK. Malignant pleural mesothelioma: an update on diagnosis and treatment options. Ther Adv Respir Dis. 2016;10:275-88.
6. Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21:2636-44.
7. Janes SM, Alrifai D, Fennell DA. Perspectives on the treatment of malignant pleural mesothelioma. N Engl J Med. 2021;385:1207-18.
8. Bertin B, Zugman M, Schvartsman G. The current treatment landscape of malignant pleural mesothelioma and future directions. Cancers. 2023;15:5808.
9. Felip E, Popat S, Dafni U, et al.; ETOP 13-18 BEAT-meso Collaborators. A randomised phase III study of bevacizumab and carboplatin-pemetrexed chemotherapy with or without atezolizumab as first-line treatment for advanced pleural mesothelioma: results of the ETOP 13-18 BEAT-meso trial. Ann Oncol. 2025;36:548-60.
10. Giri S, Lamichhane G, Pandey J, et al. Immune modulation and immunotherapy in solid tumors: mechanisms of resistance and potential therapeutic strategies. Int J Mol Sci. 2025;26:2923.
11. Abbott DM, Bortolotto C, Benvenuti S, Lancia A, Filippi AR, Stella GM. Malignant pleural mesothelioma: genetic and microenviromental heterogeneity as an unexpected reading frame and therapeutic challenge. Cancers. 2020;12:1186.
12. Chu YD, Lai MW, Yeh CT. Unlocking the potential of arginine deprivation therapy: recent breakthroughs and promising future for cancer treatment. Int J Mol Sci. 2023;24:10668.
13. National Institute for Health and Care Excellence. Nivolumab with ipilimumab for untreated unresectable malignant pleural mesothelioma. Available from: https://www.nice.org.uk/guidance/ta818. [Last accessed on 16 Mar 2026].
14. Arimura K, Hiroshima K, Nagashima Y, et al. LAG3 is an independent prognostic biomarker and potential target for immune checkpoint inhibitors in malignant pleural mesothelioma: a retrospective study. BMC Cancer. 2023;23:1206.
15. Chen S, Yu W, Shao S, et al. Establishment of predictive nomogram and web-based survival risk calculator for malignant pleural mesothelioma: a SEER database analysis. Front Oncol. 2022;12:1027149.
16. Field GC, Pavlyk I, Szlosarek PW. Bench-to-bedside studies of arginine deprivation in cancer. Molecules. 2023;28:2150.
17. He J, Xu S, Pan H, Li S, He J. Does size matter? - A population-based analysis of malignant pleural mesothelioma. Transl Lung Cancer Res. 2020;9:1041-52.
18. Urso L, Cavallari I, Sharova E, Ciccarese F, Pasello G, Ciminale V. Metabolic rewiring and redox alterations in malignant pleural mesothelioma. Br J Cancer. 2020;122:52-61.
19. Andujar P, Lacourt A, Brochard P, Pairon JC, Jaurand MC, Jean D. Five years update on relationships between malignant pleural mesothelioma and exposure to asbestos and other elongated mineral particles. J Toxicol Environ Health B Crit Rev. 2016;19:151-72.
20. Chung CTS, Da Cunha Santos G, Hwang DM, et al. FISH assay development for the detection of p16/CDKN2A deletion in malignant pleural mesothelioma. J Clin Pathol. 2010;63:630-4.
21. Felley-Bosco E, MacFarlane M. Asbestos: modern insights for toxicology in the era of engineered nanomaterials. Chem Res Toxicol. 2018;31:994-1008.
22. Feun LG, Kuo MT, Savaraj N. Arginine deprivation in cancer therapy. Curr Opin Clin Nutr Metab Care. 2015;18:78-82.
23. Cvetković Z, Marković O, Marinković G, Pejić S, Vučić V. Tumor microenvironment, inflammation, and inflammatory prognostic indices in diffuse large B-cell lymphomas: a narrative review. Int J Mol Sci. 2025;26:5670.
24. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162-74.
25. Barnett SE, Kenyani J, Tripari M, et al. BAP1 loss is associated with higher ASS1 expression in epithelioid mesothelioma: implications for therapeutic stratification. Mol Cancer Res. 2023;21:411-27.
26. Calabrò L, Bronte G, Grosso F, et al. Immunotherapy of mesothelioma: the evolving change of a long-standing therapeutic dream. Front Immunol. 2023;14:1333661.
27. Perrino M, De Vincenzo F, Cordua N, et al. Immunotherapy with immune checkpoint inhibitors and predictive biomarkers in malignant mesothelioma: work still in progress. Front Immunol. 2023;14:1121557.
28. Bertino P, Premeaux TA, Fujita T, et al. Targeting the C-terminus of galectin-9 induces mesothelioma apoptosis and M2 macrophage depletion. Oncoimmunology. 2019;8:1601482.
29. Çakan E, Lara OD, Szymanowska A, et al. Therapeutic antisense oligonucleotides in oncology: from bench to bedside. Cancers. 2024;16:2940.
30. Janjigian YY, Shitara K, Moehler M, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398:27-40.
31. Meirson T, Pentimalli F, Cerza F, et al. Comparison of 3 randomized clinical trials of frontline therapies for malignant pleural mesothelioma. JAMA Netw Open. 2022;5:e221490.
32. López-Castro R, Fuentes-Martín Á, Medina Del Valle A, et al. Advances in immunotherapy for malignant pleural mesothelioma: from emerging strategies to translational insights. Open Respir Arch. 2024;6:100323.
33. Neilly MDJ, Pearson J, Thu AW, MacRae C, Blyth KG. Contemporary management of mesothelioma. Breathe. 2024;20:230175.
34. Scherpereel A, Mazieres J, Greillier L, et al.; French Cooperative Thoracic Intergroup. Nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): a multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol. 2019;20:239-53.
35. Peters S, Scherpereel A, Cornelissen R, et al. First-line nivolumab plus ipilimumab versus chemotherapy in patients with unresectable malignant pleural mesothelioma: 3-year outcomes from CheckMate 743. Ann Oncol. 2022;33:488-99.
36. Szlosarek PW, Creelan BC, Sarkodie T, et al.; ATOMIC-Meso Study Group. Pegargiminase plus first-line chemotherapy in patients with nonepithelioid pleural mesothelioma: the ATOMIC-Meso randomized clinical trial. JAMA Oncol. 2024;10:475-83.
37. Nakajima EC, Vellanki PJ, Larkins E, et al. FDA approval summary: nivolumab in combination with ipilimumab for the treatment of unresectable malignant pleural mesothelioma. Clin Cancer Res. 2022;28:446-51.
38. Kim SS, Xu S, Cui J, et al. Histone deacetylase inhibition is synthetically lethal with arginine deprivation in pancreatic cancers with low argininosuccinate synthetase 1 expression. Theranostics. 2020;10:829-40.
39. Pirker C, Bilecz A, Grusch M, et al. Telomerase reverse transcriptase promoter mutations identify a genomically defined and highly aggressive human pleural mesothelioma subgroup. Clin Cancer Res. 2020;26:3819-30.
40. Menis J, Pasello G, Remon J. Immunotherapy in malignant pleural mesothelioma: a review of literature data. Transl Lung Cancer Res. 2021;10:2988-3000.
41. Chu CY, Lee YC, Hsieh CH, et al. Genome-wide CRISPR/Cas9 knockout screening uncovers a novel inflammatory pathway critical for resistance to arginine-deprivation therapy. Theranostics. 2021;11:3624-41.
42. Qualiotto AN, Baldavira CM, Balancin M, Ab’Saber A, Takagaki T, Capelozzi VL. Mesothelin expression remodeled the immune-matrix tumor microenvironment predicting the risk of death in patients with malignant pleural mesothelioma. Front Immunol. 2023;14:1268927.
43. Cakiroglu E, Senturk S. Genomics and functional genomics of malignant pleural mesothelioma. Int J Mol Sci. 2020;21:6342.
44. Ye LL, Peng WB, Niu YR, et al. Accumulation of TNFR2-expressing regulatory T cells in malignant pleural effusion of lung cancer patients is associated with poor prognosis. Ann Transl Med. 2020;8:1647.
45. Harber J, Kamata T, Pritchard C, Fennell D. Matter of TIME: the tumor-immune microenvironment of mesothelioma and implications for checkpoint blockade efficacy. J Immunother Cancer. 2021;9:e003032.
46. Quetel L, Meiller C, Assié JB, et al. Genetic alterations of malignant pleural mesothelioma: association with tumor heterogeneity and overall survival. Mol Oncol. 2020;14:1207-23.
47. Hegmans JP, Hemmes A, Hammad H, Boon L, Hoogsteden HC, Lambrecht BN. Mesothelioma environment comprises cytokines and T-regulatory cells that suppress immune responses. Eur Respir J. 2006;27:1086-95.
48. Haas AR, Sterman DH. Novel intrapleural therapies for malignant diseases. Respiration. 2012;83:277-92.
49. Klampatsa A, Haas AR, Moon EK, Albelda SM. Chimeric antigen receptor (CAR) T cell therapy for malignant pleural mesothelioma (MPM). Cancers. 2017;9:115.
50. Chu GJ, van Zandwijk N, Rasko JEJ. The immune microenvironment in mesothelioma: mechanisms of resistance to immunotherapy. Front Oncol. 2019;9:1366.
51. Sayan M, Mossman BT. The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Part Fibre Toxicol. 2016;13:51.
52. Waldron TJ, Quatromoni JG, Karakasheva TA, Singhal S, Rustgi AK. Myeloid derived suppressor cells: targets for therapy. Oncoimmunology. 2013;2:e24117.
53. Barbone D, Van Dam L, Follo C, et al. Analysis of gene expression in 3D spheroids highlights a survival role for ASS1 in mesothelioma. PLoS ONE. 2016;11:e0150044.
54. Markowitz P, Patel M, Groisberg R, et al. Genomic characterization of malignant pleural mesothelioma and associated clinical outcomes. Cancer Treat Res Commun. 2020;25:100232.
55. Szlosarek PW, Luong P, Phillips MM, et al. Metabolic response to pegylated arginine deiminase in mesothelioma with promoter methylation of argininosuccinate synthetase. J Clin Oncol. 2013;31:e111-3.
56. Sneddon S, Rive CM, Ma S, et al. Identification of a CD8+ T-cell response to a predicted neoantigen in malignant mesothelioma. Oncoimmunology. 2020;9:1684713.
57. Wijdeven RH, Luk SJ, Schoufour TAW, et al. Balanced epigenetic regulation of MHC class I expression in tumor cells by the histone ubiquitin modifiers BAP1 and PCGF1. J Immunol. 2024;212:446-54.
59. Xu D, Yang H, Schmid RA, Peng RW. Therapeutic landscape of malignant pleural mesothelioma: collateral vulnerabilities and evolutionary dependencies in the spotlight. Front Oncol. 2020;10:579464.
61. Kroeger B, Manning SA, Mohan V, et al. Hippo signaling regulates the nuclear behavior and DNA binding times of YAP and TEAD to control transcription. Sci Adv. 2025;11:eadw4974.
62. Guarnaccia AD, Hagenbeek TJ, Lee W, et al. TEAD-targeting small molecules induce a cofactor switch to regulate the Hippo pathway. Proc Natl Acad Sci U S A. 2025;122:e2425984122.
63. Sato K, Faraji F, Cervantes-Villagrana RD, et al. Targeting YAP/TAZ-TEAD signaling as a therapeutic approach in head and neck squamous cell carcinoma. Cancer Lett. 2025;612:217467.
64. Agioti S, Zaravinos A. Immune cytolytic activity and strategies for therapeutic treatment. Int J Mol Sci. 2024;25:3624.
65. Luo J, Xiang X, Gong G, Jiang L. Cancer-associated fibroblast-mediated immune evasion: molecular mechanisms of stromal-immune crosstalk in the tumor microenvironment. Front Immunol. 2025;16:1617662.
66. Tanimura K, Yamada T, Omura A, et al. The impact of VEGF inhibition on clinical outcomes in patients with advanced non-small cell lung cancer treated with immunotherapy: a retrospective cohort study. Front Oncol. 2021;11:663612.
67. Hu Q, Nonaka K, Wakiyama H, et al. Cytolytic activity score as a biomarker for antitumor immunity and clinical outcome in patients with gastric cancer. Cancer Med. 2021;10:3129-38.
68. Mori T, Tanaka H, Suzuki S, et al. Tertiary lymphoid structures show infiltration of effective tumor-resident T cells in gastric cancer. Cancer Sci. 2021;112:1746-57.
69. Yoshimoto A, Kasahara K, Saito K, Fujimura M, Nakao S. Granulocyte colony-stimulating factor-producing malignant pleural mesothelioma with the expression of other cytokines. Int J Clin Oncol. 2005;10:58-62.
70. Yang C, You J, Wang Y, et al. TLS and immune cell profiling: immunomodulatory effects of immunochemotherapy on tumor microenvironment in resectable stage III NSCLC. Front Immunol. 2024;15:1499731.
71. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187-94.
72. Marcq E, Waele J, Audenaerde JV, et al. Abundant expression of TIM-3, LAG-3, PD-1 and PD-L1 as immunotherapy checkpoint targets in effusions of mesothelioma patients. Oncotarget. 2017;8:89722-35.
73. Rovers S, Janssens A, Raskin J, et al. Recent advances of immune checkpoint inhibition and potential for (combined) TIGIT blockade as a new strategy for malignant pleural mesothelioma. Biomedicines. 2022;10:673.
74. Ruoff C, Mitchell A, Mondal P, et al. Resistance signatures manifested in early drug response across cancer types and species. Cancer Drug Resist. 2025;8:44.
75. Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.
76. Osmanbeyoglu HU, Palmer D, Sagan A, Sementino E, Becich MJ, Testa JR. Isolated BAP1 genomic alteration in malignant pleural mesothelioma predicts distinct immunogenicity with implications for immunotherapeutic response. Cancers. 2022;14:5626.
77. Felley-Bosco E, Stahel R. Hippo/YAP pathway for targeted therapy. Transl Lung Cancer Res. 2014;3:75-83.
78. Zhang W, Wu X, Wu L, Zhang W, Zhao X. Advances in the diagnosis, treatment and prognosis of malignant pleural mesothelioma. Ann Transl Med. 2015;3:182.
79. Zhai K, Xie R, Ru K, Zhao M. Tertiary lymphoid structures correlate with the therapeutic efficacy and prognosis of resectable esophageal squamous cell carcinoma undergoing neoadjuvant chemoradiotherapy plus immunotherapy. Front Immunol. 2025;16:1616247.
80. Locke M, Ghazaly E, Freitas MO, et al. Inhibition of the polyamine synthesis pathway is synthetically lethal with loss of argininosuccinate synthase 1. Cell Rep. 2016;16:1604-13.
81. Zhai K, Ma Y, Gao X, Ru K, Zhao M. Tertiary lymphoid structures in esophageal cancer: a novel target for immunotherapy. Front Immunol. 2025;16:1543322.
82. Alamri AM, Assiri AA, Khan B, Khan NU. Next-generation oncology: integrative therapeutic frontiers at the crossroads of precision genomics, immuno-engineering, and tumor microenvironment modulation. Med Oncol. 2025;42:482.
83. Su C, Chen S, He X, et al. Increased IgG4 expression within tertiary lymphoid structures of esophageal cancer and implications for prognosis. Front Immunol. 2025;16:1654655.
84. Bonsignore G, Ranzato E, Martinotti S. Unraveling BOLD-100 synergistic potential in pleural mesothelioma treatment: an in vitro study. Investig New Drugs. 2025;43:634-45.
85. Li W, Zhang M, Cai S, et al. A deep learning-based model (DeepMPM) to help predict survival in patients with malignant pleural mesothelioma. Transl Cancer Res. 2023;12:2887-97.
86. Szlosarek PW, Steele JP, Nolan L, et al. Arginine deprivation with pegylated arginine deiminase in patients with argininosuccinate synthetase 1-deficient malignant pleural mesothelioma: a randomized clinical trial. JAMA Oncol. 2017;3:58-66.
87. Roznovan CN, Măruțescu LG, Gradisteanu Pircalabioru G. Immuno-oncology at the crossroads: confronting challenges in the quest for effective cancer therapies. Int J Mol Sci. 2025;26:6177.
88. Di Blasio S, Wortel IM, van Bladel DA, et al. Human CD1c+ DCs are critical cellular mediators of immune responses induced by immunogenic cell death. Oncoimmunology. 2016;5:e1192739.
89. Wang YJ, Fletcher R, Yu J, Zhang L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018;5:194-203.
90. Fucikova J, Kepp O, Kasikova L, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020;11:1013.
91. Kozai H, Ogino H, Mitsuhashi A, et al. Potential of fluoropyrimidine to be an immunologically optimal partner of immune checkpoint inhibitors through inducing immunogenic cell death for thoracic malignancies. Thorac Cancer. 2024;15:369-78.
92. Sakakibara K, Sato T, Kufe DW, VonHoff DD, Kawabe T. CBP501 induces immunogenic tumor cell death and CD8 T cell infiltration into tumors in combination with platinum, and increases the efficacy of immune checkpoint inhibitors against tumors in mice. Oncotarget. 2017;8:78277-88.
93. Garg AD, More S, Rufo N, et al. Trial watch: immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology. 2017;6:e1386829.
94. Wong DY, Ong WW, Ang WH. Induction of immunogenic cell death by chemotherapeutic platinum complexes. Angew Chem Int Ed Engl. 2015;54:6483-7.
95. Sbidian E, Chaimani A, Garcia-Doval I, et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database Syst Rev. 2021;4:CD011535.
96. Chu Q, Perrone F, Greillier L, et al. Pembrolizumab plus chemotherapy versus chemotherapy in untreated advanced pleural mesothelioma in Canada, Italy, and France: a phase 3, open-label, randomised controlled trial. Lancet. 2023;402:2295-306.
97. Aerts JG, Belderbos R, Baas P, et al.; DENIM team. Dendritic cells loaded with allogeneic tumour cell lysate plus best supportive care versus best supportive care alone in patients with pleural mesothelioma as maintenance therapy after chemotherapy (DENIM): a multicentre, open-label, randomised, phase 2/3 study. Lancet Oncol. 2024;25:865-78.
98. Nowak AK, Lesterhuis WJ, Kok PS, et al. Durvalumab with first-line chemotherapy in previously untreated malignant pleural mesothelioma (DREAM): a multicentre, single-arm, phase 2 trial with a safety run-in. Lancet Oncol. 2020;21:1213-23.
99. Forde PM, Anagnostou V, Sun Z, et al. Durvalumab with platinum-pemetrexed for unresectable pleural mesothelioma: survival, genomic and immunologic analyses from the phase 2 PrE0505 trial. Nat Med. 2021;27:1910-20.
100. Pal RS, Wahlang J, Pal Y, Chaitanya M, Saxena S. Precision oncology: transforming cancer care through personalized medicine. Med Oncol. 2025;42:246.
101. Patole VP, Shinde NV, Chandak SM, Satalkar AT. Cancer research in the 21st Century: recent advances and future perspectives. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2025;41:e20250018.
102. Robinson BWS, Redwood AJ, Creaney J. How our continuing studies of the pre-clinical inbred mouse models of mesothelioma have influenced the development of new therapies. Front Pharmacol. 2022;13:858557.
103. Douma LH, van der Noort V, Lalezari F, et al. Pembrolizumab plus lenvatinib as second-line treatment in patients with pleural mesothelioma (PEMMELA): cohort 2 of a single-arm, phase 2 study. Lancet Oncol. 2025;26:1676-84.
104. Douma LH, Lalezari F, van der Noort V, et al. Pembrolizumab plus lenvatinib in second-line and third-line patients with pleural mesothelioma (PEMMELA): a single-arm phase 2 study. Lancet Oncol. 2023;24:1219-28.
105. Popat S, Curioni-Fontecedro A, Dafni U, et al. A multicentre randomised phase III trial comparing pembrolizumab versus single-agent chemotherapy for advanced pre-treated malignant pleural mesothelioma: the European Thoracic Oncology Platform (ETOP 9-15) PROMISE-meso trial. Ann Oncol. 2020;31:1734-45.
106. Voron T, Colussi O, Marcheteau E, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212:139-48.
107. Lee WS, Yang H, Chon HJ, Kim C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med. 2020;52:1475-85.
108. Huang Y, Yuan J, Righi E, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109:17561-6.
109. Chiec L, Bruno DS. Immunotherapy for treatment of pleural mesothelioma: current and emerging therapeutic strategies. Int J Mol Sci. 2024;25:10861.
110. Rovers S, Van Audenaerde J, Verloy R, et al. Co-targeting of VEGFR2 and PD-L1 promotes survival and vasculature normalization in pleural mesothelioma. Oncoimmunology. 2025;14:2512104.
111. Magkouta SF, Vaitsi PC, Pappas AG, et al. CSF1/CSF1R axis blockade limits mesothelioma and enhances efficiency of anti-PDL1 immunotherapy. Cancers. 2021;13:2546.
112. Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. Pegylated arginine deiminase (ADI-SS PEG20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer Res. 2002;62:5443-50.
113. Adusumilli PS, Zauderer MG, Rivière I, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab. Cancer Discov. 2021;11:2748-63.
114. Davis A, Ke H, Kao S, Pavlakis N. An update on emerging therapeutic options for malignant pleural mesothelioma. Lung Cancer. 2022;13:1-12.
115. Klampatsa A, Albelda SM. Current advances in CAR T cell therapy for malignant mesothelioma. J Cell Immunol. 2020;2:192-200.
116. Haas AR, Tanyi JL, O’Hara MH, et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol Ther. 2019;27:1919-29.
117. Haas AR, Golden RJ, Litzky LA, et al. Two cases of severe pulmonary toxicity from highly active mesothelin-directed CAR T cells. Mol Ther. 2023;31:2309-25.
118. Chen Q, Lu L, Ma W. Efficacy, safety, and challenges of CAR T-cells in the treatment of solid tumors. Cancers. 2022;14:5983.
119. Hiltbrunner S, Britschgi C, Schuberth P, et al. Local delivery of CAR T cells targeting fibroblast activation protein is safe in patients with pleural mesothelioma: first report of FAPME, a phase I clinical trial. Ann Oncol. 2021;32:120-1.
120. Astarita JL, Acton SE, Turley SJ. Podoplanin: emerging functions in development, the immune system, and cancer. Front Immunol. 2012;3:283.
121. Krishnan H, Rayes J, Miyashita T, et al. Podoplanin: an emerging cancer biomarker and therapeutic target. Cancer Sci. 2018;109:1292-9.
122. Huang Z, Dewanjee S, Chakraborty P, et al. CAR T cells: engineered immune cells to treat brain cancers and beyond. Mol Cancer. 2023;22:22.
123. Peng L, Sferruzza G, Yang L, Zhou L, Chen S. CAR-T and CAR-NK as cellular cancer immunotherapy for solid tumors. Cell Mol Immunol. 2024;21:1089-108.
124. Pan K, Farrukh H, Chittepu VCSR, Xu H, Pan CX, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022;41:119.
125. Sun X, Zhao P, Lin J, Chen K, Shen J. Recent advances in access to overcome cancer drug resistance by nanocarrier drug delivery system. Cancer Drug Resist. 2023;6:390-415.
126. McCarthy D, Lofgren M, Watt A, et al. Functional enhancement of mesothelin-targeted TRuC-T cells by a PD1-CD28 chimeric switch receptor. Cancer Immunol Immunother. 2023;72:4195-207.
127. Sun Y, Yang H, Xu Q, et al. Anti-PD-1 nanobody-armored MSLN CAR-T therapy for malignant mesothelioma: preclinical and clinical studies. Adv Sci. ;2025:e08754.
128. Liu S, Li X, Wei W, et al. Translational selenium nanoparticles enhance NKG2D-mediated cytotoxicity of NK cells against malignant pleural mesothelioma cells through the TrxR1-pSTAT3 pathway. Nano Today. 2025;62:102720.
129. Kong X, Zhang J, Chen S, et al. Immune checkpoint inhibitors: breakthroughs in cancer treatment. Cancer Biol Med. 2024;21:451-72.
130. Kutle I, Polten R, Stalp JL, et al. Anti-mesothelin CAR-NK cells as a novel targeted therapy against cervical cancer. Front Immunol. 2024;15:1485461.
131. Jung D, Choi E, Jeoung YH, et al. Nanobody-based CAR NK cells for possible immunotherapy of mesothelin+ tumors. Immune Netw. 2025;25:e23.
132. Castelletti L, Yeo D, van Zandwijk N, Rasko JEJ. Anti-mesothelin CAR T cell therapy for malignant mesothelioma. Biomark Res. 2021;9:11.
133. Tano ZE, Chintala NK, Li X, Adusumilli PS. Novel immunotherapy clinical trials in malignant pleural mesothelioma. Ann Transl Med. 2017;5:245.
134. Hegmans JP, Veltman JD, Lambers ME, et al. Consolidative dendritic cell-based immunotherapy elicits cytotoxicity against malignant mesothelioma. Am J Respir Crit Care Med. 2010;181:1383-90.
135. Van den Bossche J, De Laere M, Deschepper K, et al. Integration of the PD-L1 inhibitor atezolizumab and WT1/DC vaccination into standard-of-care first-line treatment for patients with epithelioid malignant pleural mesothelioma-Protocol of the Immuno-MESODEC study. PLoS One. 2024;19:e0307204.
136. Terenziani R, Zoppi S, Fumarola C, Alfieri R, Bonelli M. Immunotherapeutic approaches in malignant pleural mesothelioma. Cancers. 2021;13:2793.
137. Garcia-Carbonero R, Paz-Ares L. Systemic chemotherapy in the management of malignant peritoneal mesothelioma. Eur J Surg Oncol. 2006;32:676-81.
138. Obacz J, Yung H, Shamseddin M, et al. Biological basis for novel mesothelioma therapies. Br J Cancer. 2021;125:1039-55.
139. Nicolini F, Bocchini M, Bronte G, et al. Malignant pleural mesothelioma: state-of-the-art on current therapies and promises for the future. Front Oncol. 2019;9:1519.
140. Dozier J, Zheng H, Adusumilli PS. Immunotherapy for malignant pleural mesothelioma: current status and future directions. Transl Lung Cancer Res. 2017;6:315-24.
141. Banerji S, Meyers DE, Harlos C, Dawe DE. The role of immunotherapy in the treatment of malignant pleural mesothelioma. Curr Oncol. 2021;28:4542-51.
142. Gray SG. Emerging avenues in immunotherapy for the management of malignant pleural mesothelioma. BMC Pulm Med. 2021;21:148.
143. Zolaly MA, Mahallawi W, Khawaji ZY, Alahmadi MA. The clinical advances of oncolytic viruses in cancer immunotherapy. Cureus. 2023;15:e40742.
144. Ponce S, Cedrés S, Ricordel C, et al. ONCOS-102 plus pemetrexed and platinum chemotherapy in malignant pleural mesothelioma: a randomized phase 2 study investigating clinical outcomes and the tumor microenvironment. J Immunother Cancer. 2023;11:e007552.
145. Kuryk L, Møller AW, Jaderberg M. Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. Oncoimmunology. 2019;8:e1532763.
146. Monberg TJ, Pakola SA, Albieri B, et al. Safety and efficacy of combined treatment with tumor-infiltrating lymphocytes and oncolytic adenovirus TILT-123 in metastatic melanoma. Cell Rep Med. 2025;6:102016.
147. Mondal M, Guo J, He P, Zhou D. Recent advances of oncolytic virus in cancer therapy. Hum Vaccin Immunother. 2020;16:2389-402.
148. Gray SG, Mutti L. Immunotherapy for mesothelioma: a critical review of current clinical trials and future perspectives. Transl Lung Cancer Res. 2020;9 Suppl:S100-19.
149. Omolekan TO, Folahan JT, Tesfay MZ, et al. Viral warfare: unleashing engineered oncolytic viruses to outsmart cancer’s defenses. Front Immunol. 2025;16:1618751.
150. Cheng C, Wang Q, Zhang S. Synergy of oncolytic adenovirus and immune checkpoint inhibitors: transforming cancer immunotherapy paradigms. Front Immunol. 2025;16:1610858.
151. Heiniö C, Clubb J, Kudling T, et al. Effective combination immunotherapy with oncolytic adenovirus and anti-PD-1 for treatment of human and murine ovarian cancers. Diseases. 2022;10:52.
152. Torricelli F, Donati B, Manicardi V, et al. CDKN2A deletion is associated with immune desertification in diffuse pleural mesothelioma. J Exp Clin Cancer Res. 2025;44:256.
153. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30:36-50.
154. Carbotti G, Dozin B, Martini S, et al. IL-27 mediates PD-L1 expression and release by human mesothelioma cells. Cancers. 2021;13:4011.
155. Ahmadzada T, Lee K, Clarke C, et al. High BIN1 expression has a favorable prognosis in malignant pleural mesothelioma and is associated with tumor infiltrating lymphocytes. Lung Cancer. 2019;130:35-41.
156. Li X, Xiang Y, Li F, Yin C, Li B, Ke X. WNT/β-catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front Immunol. 2019;10:2293.
157. Zhong Z, Virshup DM. Wnt signaling and drug resistance in cancer. Mol Pharmacol. 2020;97:72-89.
158. Pai SG, Carneiro BA, Mota JM, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017;10:101.
159. Lecarpentier Y, Schussler O, Hébert JL, Vallée A. Multiple targets of the canonical WNT/β-catenin signaling in cancers. Front Oncol. 2019;9:1248.
160. Vogl M, Rosenmayr A, Bohanes T, et al. Biomarkers for malignant pleural mesothelioma - a novel view on inflammation. Cancers. 2021;13:658.
161. Caja L, Dituri F, Mancarella S, et al. TGF-β and the tissue microenvironment: relevance in fibrosis and cancer. Int J Mol Sci. 2018;19:1294.
162. Czajka-Francuz P, Prendes MJ, Mankan A, et al. Mechanisms of immune modulation in the tumor microenvironment and implications for targeted therapy. Front Oncol. 2023;13:1200646.
163. Ghahremanifard P, Chanda A, Bonni S, Bose P. TGF-β mediated immune evasion in cancer-spotlight on cancer-associated fibroblasts. Cancers. 2020;12:3650.
164. Fatima S. Tumor microenvironment: a complex landscape of cancer development and drug resistance. Cureus. 2025;17:e82090.
165. Shin MH, Oh E, Kim Y, et al. Recent advances in CAR-based solid tumor immunotherapy. Cells. 2023;12:1606.
166. Lee HS, Jang HJ, Ramineni M, et al. A phase II window of opportunity study of neoadjuvant PD-L1 versus PD-L1 plus CTLA-4 blockade for patients with malignant pleural mesothelioma. Clin Cancer Res. 2023;29:548-59.
167. Ghafoor A, Hassan R. Somatic BAP1 loss as a predictive biomarker of overall survival in patients with malignant pleural mesothelioma treated with chemotherapy. J Thorac Oncol. 2022;17:862-4.
168. Righi L, Duregon E, Vatrano S, et al. BRCA1-associated protein 1 (BAP1) immunohistochemical expression as a diagnostic tool in malignant pleural mesothelioma classification: a large retrospective study. J Thorac Oncol. 2016;11:2006-17.
169. Pinton G, Wang Z, Balzano C, et al. CDKN2A determines mesothelioma cell fate to EZH2 inhibition. Front Oncol. 2021;11:678447.
170. Gutierrez-Sainz L, Cruz P, Martinez-Recio S, et al. Malignant pleural mesothelioma: clinical experience and prognostic value of derived neutrophil-to-lymphocyte ratio and PD-L1 expression. Clin Transl Oncol. 2021;23:2030-5.
171. Baas P, Scherpereel A, Nowak AK, et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet. 2021;397:375-86.
172. Sun B, Dong Y, Xu J, Wang Z. Current status and progress in immunotherapy for malignant pleural mesothelioma. Chronic Dis Transl Med. 2022;8:91-9.
173. Popat S, Baas P, Faivre-Finn C, et al.; ESMO Guidelines Committee. Electronic address: [email protected]. Malignant pleural mesothelioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2022;33:129-42.
174. Wang Q, Xu C, Wang W, et al. Chinese expert consensus on the diagnosis and treatment of malignant pleural mesothelioma. Thorac Cancer. 2023;14:2715-31.
175. Mansfield AS, Symanowski JT, Peikert T. Systematic review of response rates of sarcomatoid malignant pleural mesotheliomas in clinical trials. Lung Cancer. 2014;86:133-6.
176. Boutros CS, Risa EL, Jiang B, et al. Different diseases, different outcomes: a comparative study of peritoneal and pleural subtypes. J Surg Res. 2025;312:111-8.
177. Chintala NK, Choe JK, McGee E, et al. Correlative analysis from a phase I clinical trial of intrapleural administration of oncolytic vaccinia virus (Olvi-vec) in patients with malignant pleural mesothelioma. Front Immunol. 2023;14:1112960.
178. Illei PB, Rusch VW, Zakowski MF, Ladanyi M. Homozygous deletion of CDKN2A and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesotheliomas. Clin Cancer Res. 2003;9:2108-13.
179. Hong SH. Hippo pathway as another oncogenic mediator to promote immune evasion by PD-L1 signaling. J Thorac Dis. 2019;11 Suppl:S318-21.
180. Yang H, Hall SRR, Sun B, et al. NF2 and canonical Hippo-YAP pathway define distinct tumor subsets characterized by different immune deficiency and treatment implications in human pleural mesothelioma. Cancers. 2021;13:1561.
181. Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819-29.
182. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231-5.
183. Yoshikawa Y, Kuribayashi K, Minami T, Ohmuraya M, Kijima T. Epigenetic alterations and biomarkers for immune checkpoint inhibitors-current standards and future perspectives in malignant pleural mesothelioma treatment. Front Oncol. 2020;10:554570.
184. Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249-53.
185. Bhat KP, Ümit Kaniskan H, Jin J, Gozani O. Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat Rev Drug Discov. 2021;20:265-86.
186. Rugo HS, Jacobs I, Sharma S, et al. The promise for histone methyltransferase inhibitors for epigenetic therapy in clinical oncology: a narrative review. Adv Ther. 2020;37:3059-82.
187. Khadeir R, Szyszko T, Szlosarek PW. Optimizing arginine deprivation for hard-to-treat cancers. Oncotarget. 2017;8:96468-9.
188. Chintala NK, Restle D, Quach H, et al. CAR T-cell therapy for pleural mesothelioma: rationale, preclinical development, and clinical trials. Lung Cancer. 2021;157:48-59.
189. Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H, Sánchez-García FJ. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol. 2016;7:52.
190. Al-Taei S, Salimu J, Spary LK, Clayton A, Lester JF, Tabi Z. Prostaglandin E2-mediated adenosinergic effects on CD14+ cells: self-amplifying immunosuppression in cancer. Oncoimmunology. 2017;6:e1268308.
191. Cersosimo F, Barbarino M, Lonardi S, et al. Mesothelioma malignancy and the microenvironment: molecular mechanisms. Cancers. 2021;13:5664.
192. Russo A, Incorvaia L, Del Re M, et al. The molecular profiling of solid tumors by liquid biopsy: a position paper of the AIOM-SIAPEC-IAP-SIBioC-SIC-SIF Italian Scientific Societies. ESMO Open. 2021;6:100164.
193. Gerber B, Manzoni M, Spina V, et al. Circulating tumor DNA as a liquid biopsy in plasma cell dyscrasias. Haematologica. 2018;103:e245-8.
194. Desai AP, Kosari F, Disselhorst M, et al. Dynamics and survival associations of T cell receptor clusters in patients with pleural mesothelioma treated with immunotherapy. J Immunother Cancer. 2023;11:e006035.
195. Zhang M, Liu C, Tu J, et al. Advances in cancer immunotherapy: historical perspectives, current developments, and future directions. Mol Cancer. 2025;24:136.
196. Shmatko A, Ghaffari Laleh N, Gerstung M, Kather JN. Artificial intelligence in histopathology: enhancing cancer research and clinical oncology. Nat Cancer. 2022;3:1026-38.
197. Courtiol P, Maussion C, Moarii M, et al. Deep learning-based classification of mesothelioma improves prediction of patient outcome. Nat Med. 2019;25:1519-25.
198. Galateau Salle F, Le Stang N, Nicholson AG, et al. New insights on diagnostic reproducibility of biphasic mesotheliomas: a multi-institutional evaluation by the international mesothelioma panel from the MESOPATH Reference Center. J Thorac Oncol. 2018;13:1189-203.
199. Marra A, Morganti S, Pareja F, et al. Artificial intelligence entering the pathology arena in oncology: current applications and future perspectives. Ann Oncol. 2025;36:712-25.
200. Zhang C, de A F Fonseca L, Shi Z, et al. Systematic review of radiomic biomarkers for predicting immune checkpoint inhibitor treatment outcomes. Methods. 2021;188:61-72.
201. Aerts HJ, Velazquez ER, Leijenaar RT, et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat Commun. 2014;5:4006.
202. Li D, Cao Y, Petrella F, Zou Y. Malignant pleural mesothelioma treated with cytoreductive video-assisted thoracic surgery plus hyperthermic intrathoracic chemotherapy: a case report. J Thorac Dis. 2024;16:8133-41.
203. Choi HY, Chang JE. Targeted therapy for cancers: from ongoing clinical trials to FDA-approved drugs. Int J Mol Sci. 2023;24:13618.
204. Ceresoli GL, Aerts JG, Dziadziuszko R, et al. Tumour treating fields in combination with pemetrexed and cisplatin or carboplatin as first-line treatment for unresectable malignant pleural mesothelioma (STELLAR): a multicentre, single-arm phase 2 trial. Lancet Oncol. 2019;20:1702-9.
205. Kutuk T, Atak E, La Rosa A, Kotecha R, Mehta MP, Chuong MD. Tumor treating fields: narrative review of a promising treatment modality for cancer. Chin Clin Oncol. 2023;12:64.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].