


Mechanism-driven therapy for diabetic cardiovascular disease
 0
0 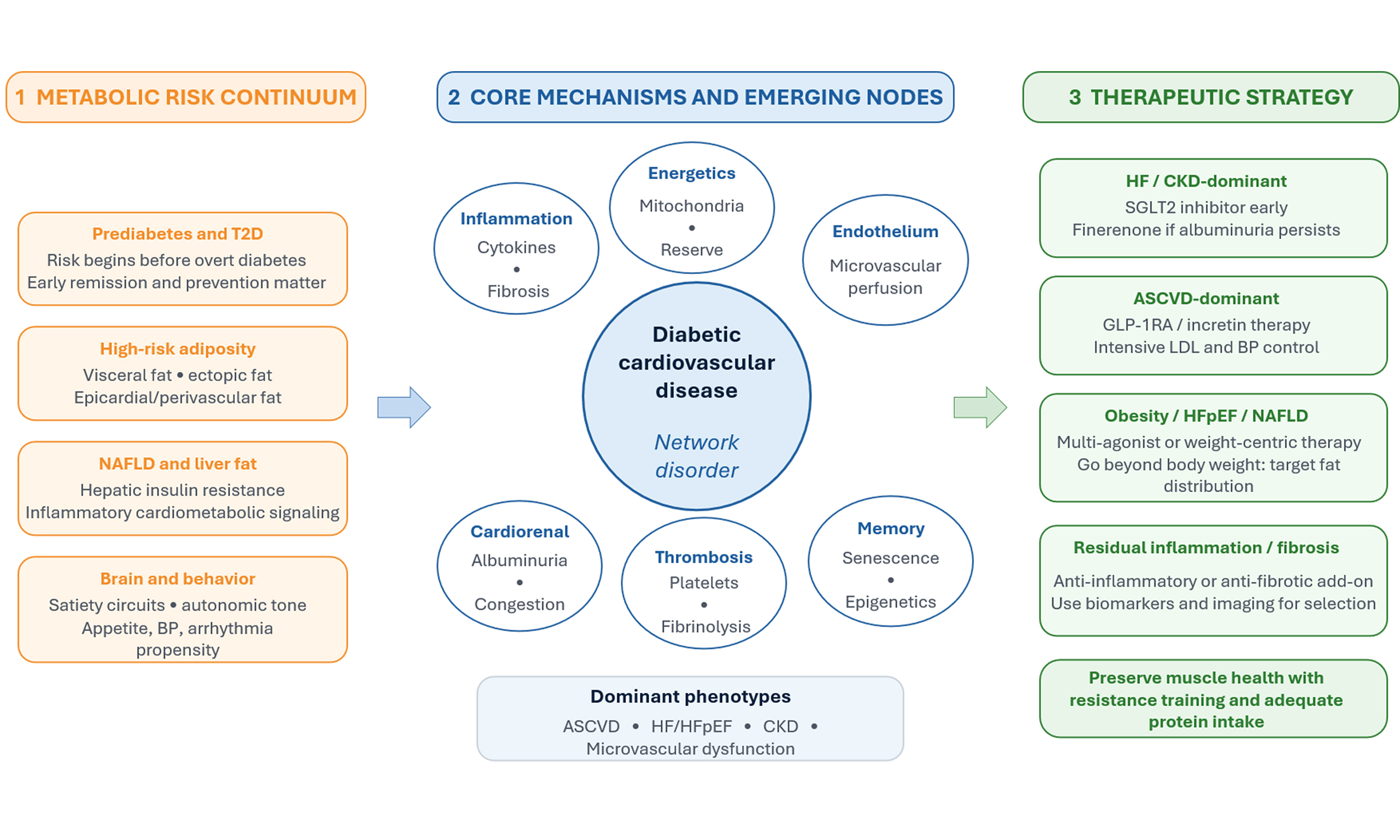
DIABETIC CARDIOVASCULAR DISEASE IS A NETWORK DISORDER, NOT A GLUCOSE-ONLY COMPLICATION
Diabetes accelerates cardiovascular (CV) target-organ damage through multiple interacting biological modules that amplify risk even when glycemia is modestly controlled[1]. These modules include (i) adipose tissue dysfunction with altered adipokine secretion and immune-cell infiltration; (ii) ectopic lipid deposition in liver, myocardium, and perivascular/epicardial fat; (iii) endothelial dysfunction with impaired nitric oxide bioavailability and heightened oxidative stress; (iv) chronic kidney disease (CKD)-mediated volume, pressure, and uremic-toxin stress; and (v) autonomic and neurohormonal activation that promotes hypertension, arrhythmias, and remodeling[1]. The clinical spectrum includes macrovascular disease as well as myocardial, renal, and microvascular complications, including heart failure (HF) with preserved ejection fraction (HFpEF), diabetic cardiomyopathy, CKD, and coronary microcirculatory dysfunction[1]. Importantly, cardiometabolic risk accrues even before overt diabetes; emerging evidence suggests that achieving remission of prediabetes is associated with lower subsequent cardiorenal and CV morbidity[2], reinforcing prevention and early intervention as core therapeutic strategies. Likewise, the distribution of body fat (visceral and ectopic depots) may be more relevant to risk modification than body weight alone[3].
CORE MECHANISMS AND EMERGING NODES CONNECTING METABOLISM TO CV INJURY
Core biological modules linking metabolism to diabetic cardiovascular disease (DCVD) include the following[4]. First, immune-metabolic coupling: insulin resistance and adipose stress skew innate and adaptive immune tone toward pro-inflammatory phenotypes, thereby sustaining cytokine signaling that destabilizes plaques and promotes myocardial fibrosis[5]. Second, myocardial energetics: diabetes shifts substrate use toward fatty acid oxidation, increasing the oxygen cost per ATP and reducing efficiency and metabolic flexibility; mitochondrial dysfunction and impaired ketone handling further contribute to the loss of contractile reserve[6]. Third, vascular biology: advanced glycation end products, oxidative stress, and impaired endothelial repair compromise vasomotor function and microvascular perfusion, thereby contributing to angina without obstructive coronary disease and to HFpEF[7]. Fourth, the cardiorenal axis: glomerular hyperfiltration, albuminuria, and tubuloglomerular feedback abnormalities link diabetes to sodium retention, congestion, and progressive remodeling[8]. Finally, thrombo-inflammatory signaling, platelet hyperreactivity, impaired fibrinolysis, and altered lipoprotein composition create a pro-atherothrombotic milieu[9].
An underappreciated yet increasingly actionable node is epicardial and perivascular adipose tissue[10]. In diabetes and obesity, these depots expand and shift toward a pro-inflammatory, pro-fibrotic secretome that acts locally on the coronary microcirculation and the adjacent myocardium[11]. This paracrine signaling can promote microvascular rarefaction, myocardial stiffness, and atrial remodeling, thereby providing a mechanistic link between adiposity and phenotypes such as HFpEF, microvascular angina, and atrial fibrillation[12].
Additional emerging mechanisms likely contribute to persistent risk despite contemporary risk-factor control. Gut-immune-metabolic crosstalk, including altered bile acid signaling and microbial-derived metabolites, may amplify vascular inflammation and insulin resistance[13]. At the tissue level, cellular senescence and epigenetic metabolic memory can sustain oxidative stress and endothelial dysfunction even after glycemic improvement, supporting the rationale for earlier intervention and therapies that directly target inflammation, fibrosis, and vascular repair[14]. Nonalcoholic fatty liver disease (NAFLD) should be viewed as both a marker and mediator of high-risk adiposity, linking hepatic insulin resistance and inflammatory signaling to atherogenesis and HFpEF risk; therapies that reduce liver fat (intensive lifestyle change and incretin-based or multi-agonist pharmacotherapy) may therefore confer benefits beyond body mass index (BMI) alone[15-18]. Finally, the brain is not merely an appetite “controller” but a cardiometabolic effector organ. Central satiety circuits and autonomic pathways shape sympathetic tone, blood pressure, propensity for arrhythmias, and cardiometabolic behavior, providing an additional mechanistic lens for incretin-based therapies[19].
THERAPEUTIC STRATEGIES ALIGNED TO THE MECHANISM: WHAT HAS CHANGED?
Outcome trials have shown that mechanism-targeted cardiometabolic therapies improve hard outcomes in patients with DCVD or related high-risk cardiometabolic phenotypes. Sodium-glucose cotransporter-2 (SGLT2) inhibitors provide rapid benefits in HF and CKD. These effects likely reflect natriuresis, reduced congestion, restoration of tubuloglomerular feedback, and improved myocardial efficiency[20]. Glucagon-like peptide-1 receptor agonists (GLP-1RAs) reduce major adverse cardiovascular disease (CVD) events, primarily in atherosclerotic phenotypes, plausibly via weight loss, blood pressure reduction, improved endothelial function, and anti-inflammatory effects[21]. Oral semaglutide has been associated with a significantly lower risk of major adverse CVD events in type 2 diabetes (T2D) and atherosclerotic CVD (ASCVD), CKD, or both, without increasing the incidence of serious adverse events[22]. The recent demonstration that an oral GLP-1RA formulation can deliver CVD benefit reinforces the importance of route of administration and adherence-friendly options in shaping population-level impact, particularly in primary care settings, where injectable therapies may face barriers[23].
A practical implication is the use of phenotype-driven sequencing rather than uniform escalation [Table 1]. In HF and CKD-dominant presentations (congestion, albuminuria, declining estimated glomerular filtration rate), prioritize early SGLT2 inhibition and consider add-on non-steroidal mineralocorticoid receptor antagonism if albuminuria persists. In ASCVD-dominant disease (prior myocardial infarction or stroke, high coronary plaque burden), prioritize incretin-based therapy alongside intensive lipid and blood pressure control. In obesity-driven cardiomyopathy or HFpEF (visceral or epicardial adiposity, exercise intolerance), weight-centric incretin or multi-agonist strategies may be foundational, paired with structured exercise to preserve lean mass.
Phenotype-guided mechanism-based treatment priorities in diabetic cardiovascular disease
| Mechanistic driver | Dominant phenotype and clinical clues | Therapeutic priority and strategy |
| Cardiorenal hemodynamics and congestion | HF (including HFpEF), CKD, albuminuria, volume sensitivity | SGLT2 inhibitor early; optimize HF and hypertension therapy; consider finerenone with albuminuria[20,36] |
| Adiposity and immune-metabolic inflammation | Obesity, visceral or epicardial fat, NAFLD, HFpEF symptoms, atrial arrhythmia tendency | Incretin-based or multi-agonist weight-centric therapy; lifestyle with resistance training; address sleep and BP[15,16,25,39] |
| Atherothrombosis and endothelial dysfunction | Established ASCVD, high plaque burden, microvascular angina | Incretin therapy plus intensive LDL and BP control; antiplatelet strategy per risk; smoking cessation[21,22,41] |
| Fibrosis and residual inflammatory risk | Elevated hsCRP or IL-6, diabetic kidney disease, imaging fibrosis, persistent risk despite guideline therapy | Finerenone where indicated; consider anti-inflammatory approaches in selected patients; monitor biomarkers[33-36] |
It should be emphasized that recent outcome trials (SELECT, SURPASS-CVOT, SOUL[22,24-26]) suggest that the CVD benefits of incretin-based therapies may not be fully explained by weight loss alone. Clearly, the development of incretin-based therapies has been a remarkable advance in the management of patients living with CVD-CKD-metabolic syndrome, including T2D and obesity. Because of their powerful effects on appetite and food craving, which lead to reduced food intake and substantial weight loss, the assumption is that the effects of incretins on weight loss are directly related to the reduction in CVD events observed in these recent trials. This has spurred the continued development of even more powerful incretin-based therapies that would produce greater weight loss, with the hope of generating further benefits in terms of protection against adverse CVD outcomes. However, there appears to be a mismatch between the degree of weight loss and the observed CVD benefits with incretin-based therapies. In a prespecified analysis of SELECT, the CV benefit of semaglutide appeared independent of weight loss, and the magnitude of weight reduction was similar to that achieved in Look AHEAD. However, Look AHEAD was an intensive lifestyle intervention trial conducted in adults with overweight or obesity and T2D, with distinct behavioral context and background medication dynamics, whereas SELECT and SURPASS-CVOT evaluated pharmacotherapy. Therefore, direct mechanistic comparisons should be made cautiously, because the biological pathways engaged by weight loss and the accompanying cardiometabolic milieu may differ meaningfully across these settings. Notably, the primary Look AHEAD analysis did not show a reduction in CV events despite greater weight loss in the intervention group, whereas SELECT showed a small association between CV benefit and reduction in waist circumference, a crude anthropometric marker of abdominal-visceral adiposity[24,25,27]. In patients with T2D, the SURPASS-CVOT trial showed that, at 36 months, the magnitude of weight loss in the tirzepatide group (-11.6%) was more than twice that in the dulaglutide group (-4.8%). Despite this substantial difference in weight loss between the two treatment arms, the primary endpoint occurred in 12.2% of patients in the tirzepatide group vs. 13.1% in the dulaglutide group [P = 0.003 for hazard ratio (HR) noninferiority and P = 0.09 for HR superiority][26]. In patients with T2D and atherosclerotic CVD, CKD, or both, the SOUL trial showed that oral semaglutide at a target dose of 14 mg daily resulted in a 14% reduction (HR 0.86; 95% confidence interval (CI): 0.77 to 0.96; P = 0.006) in the primary endpoint of major adverse CVD events (a composite of death from CVD causes, nonfatal myocardial infarction, or nonfatal stroke). However, there was only an approximately 3 kg difference in body weight loss in the semaglutide group vs. placebo (estimated difference -2.95 kg; 95%CI: -3.18 to -2.73)[22].
Thus, weight loss alone may not be an adequate metric for fully appreciating the benefits of incretin-based therapies on inflammatory and other processes that modulate CVD risk. In HFpEF, the benefits for functional capacity and quality of life are likely mediated by treating obesity, not necessarily by HFpEF itself, and improvements in N-terminal pro-B-type natriuretic peptide levels and CVD outcomes may also be driven by the incretin pathway rather than just weight loss[28,29].
Many cardiometabolic imaging studies have shown that patients with T2D have greater visceral adipose tissue (VAT) and liver fat than individuals without T2D[17,18]. As weight loss induced by diet or physical activity/exercise can also produce a significant loss of visceral/ectopic fat way beyond what could be estimated by the magnitude of weight loss[30,31], one likely scenario in the SURPASS-CVOT trial is that the energy deficit achieved in the dulaglutide arm was sufficient to induce a substantial mobilization of VAT and of liver fat that could not be fully appreciated from the magnitude of weight loss. Imaging studies measuring these hidden fat depots have reported marked reductions in VAT and liver fat with incretin-based therapies. Dedicated cardiometabolic imaging trials with incretin-based therapies will need to be conducted to confirm this hypothesis[16,32]. With the relevance of these compounds to the management of patients with high-risk adiposity phenotypes, there is an opportunity to go beyond body weight and weight loss as clinically relevant metrics/outcomes. By measuring VAT and ectopic fat depots, as well as critical behaviors and their outcomes, we may be better positioned to understand how these revolutionary compounds work and optimize their use in clinical practice.
Next-generation incretin and weight-centric therapeutics
Dual and triple incretin agonists [e.g., glucagon-like peptide-1 (GLP-1)/glucose-dependent insulinotropic polypeptide (GIP) and GLP-1/GIP/glucagon] and potent anti-obesity pharmacotherapy shift the field toward adiposity as a treatable mechanism. Beyond weight loss, reductions in VAT and ectopic fat (hepatic steatosis and epicardial fat) may reprogram inflammatory tone and improve diastolic function. Key questions for DCVD include whether a greater reduction in adiposity yields incremental plaque stabilization, whether it modifies arrhythmia burden through autonomic and atrial remodeling, and how best to sequence incretin-based agents with SGLT2 inhibitors and guideline-directed HF therapies.
Inflammation, fibrosis, and vascular repair as explicit targets
Diabetes-related CV injury exhibits a prominent inflammatory-fibrotic signature[33]. Clinical proof of concept for anti-inflammatory risk reduction in atherosclerosis [e.g., interleukin (IL)-1β pathway inhibition or low-dose colchicine strategies] raises the possibility of targeted use in ASCVD-dominant DCVD with residual inflammatory risk despite optimal lipid lowering[34,35]. In parallel, non-steroidal mineralocorticoid receptor antagonism with finerenone reduces cardiorenal events in T2D with CKD and albuminuria, supporting a broader anti-fibrotic paradigm[33,36]. Future directions include identifying biomarkers [e.g., high-sensitivity C-reactive protein (hsCRP), IL-6, albuminuria, imaging of epicardial fat or myocardial fibrosis] to guide patient matching to anti-inflammatory or anti-fibrotic intensification. From a practical standpoint, anti-inflammatory intensification is most defensible in ASCVD-dominant DCVD with evidence of residual inflammatory risk (e.g., persistently elevated hsCRP despite optimized lipid lowering) and recurrent events or high plaque burden, while anti-fibrotic intensification is most actionable in T2D with CKD and albuminuria (where finerenone is indicated) and in HFpEF/structural heart disease phenotypes where imaging or biomarkers suggest a fibrotic substrate. These phenotype cues can help avoid indiscriminate add-on therapy while trials refine precision selection.
Combination therapy, implementation, and precision cardiometabolic care
The highest-risk patients rarely have isolated disease. They typically present with overlapping ASCVD, HF, and CKD. A mechanistically rational combination therapy, including SGLT2 inhibition to improve cardiorenal hemodynamics, incretin-based therapy to reduce adiposity and inflammation, and aggressive lipid and blood pressure control, should be considered the default rather than the exception, unless limited by tolerability or other factors such as cost. The field now requires implementation science: simplifying algorithms, integrating multidisciplinary care, and assessing real-world adherence. Precision approaches may arise from (i) phenotyping (ASCVD-dominant vs. HF/CKD-dominant); (ii) biomarker stratification (inflammatory risk, albuminuria); and (iii) imaging-enabled evaluation of visceral/epicardial fat, plaque features, and myocardial fibrosis. Ultimately, the goal is to align therapy to the mechanism, delivering earlier and more durable prevention of diabetic target-organ damage.
Key implementation barriers include therapeutic inertia, fragmented cardiology-endocrinology-nephrology workflows, injection aversion, and payer-driven step therapy. Actionable solutions include simple phenotype-based algorithms embedded in electronic health records, pharmacist- or nurse-led initiation and monitoring pathways, shared cardiometabolic clinics, and structured follow-up focused on tolerability, renal function, and volume status. Certainly, strategies are needed to improve adherence, as 1-year discontinuation rates are 65% among those without T2D and 46% among those with T2D[37].
Finally, although these therapies produce marked reductions in adiposity, they may also be accompanied by reductions in lean mass. In a small study (SEMALEAN), semaglutide was associated with improved handgrip strength despite reductions in lean mass; however, this observation requires confirmation in larger, more diverse cohorts[38]. Therefore, future studies should systematically assess comprehensive muscle health (quantity, quality, and function) and physical function during incretin-based and weight-loss pharmacotherapy. As potent weight-loss pharmacotherapies continue to expand, treatment programs should incorporate resistance training and adequate protein intake to mitigate the risk of sarcopenia and functional decline[39,40].
Ultimately, DCVD should be managed through earlier, phenotype-guided, mechanism-based prevention and combination therapy rather than stepwise glucose-centric escalation after target-organ damage is established.
DECLARATIONS
Authors’ contributions
Contributed equally to the conception, drafting, revision, and final approval of the manuscript: Sanchis-Gomar F, Lavie CJ, Carbone S, Neeland IJ
Read and approved the final manuscript: Sanchis-Gomar F, Lavie CJ, Carbone S, Neeland IJ
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (OpenAI, GPT-5.2, accessed 2026-04-28) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
None.
Conflicts of interest
Lavie CJ is an Editorial Board Member of the journal Metabolism and Target Organ Damage. Lavie CJ was not involved in any steps of editorial processing, notably including reviewer selection, manuscript handling, and decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Li Y, Liu Y, Liu S, et al. Diabetic vascular diseases: molecular mechanisms and therapeutic strategies. Signal Transduct Target Ther. 2023;8:152.
2. Sandforth A, von Schwartzenberg RJ, Arreola EV, et al. Mechanisms of weight loss-induced remission in people with prediabetes: a post-hoc analysis of the randomised, controlled, multicentre Prediabetes Lifestyle Intervention Study (PLIS). Lancet Diabetes Endocrinol. 2023;11:798-810.
3. Sandforth A, Arreola EV, Hanson RL, et al. Prevention of type 2 diabetes through prediabetes remission without weight loss. Nat Med. 2025;31:3330-40.
4. Chen L, Chen M, Yang X, et al. Energy metabolism in cardiovascular diseases: unlocking the hidden powerhouse of cardiac pathophysiology. Front Endocrinol. 2025;16:1617305.
5. Karakasis P, Stachteas P, Iliakis P, et al. Inflammation and resolution in obesity-related cardiovascular disease. Int J Mol Sci. 2026;27:535.
6. Karwi QG, Sun Q, Lopaschuk GD. The contribution of cardiac fatty acid oxidation to diabetic cardiomyopathy severity. Cells. 2021;10:3259.
7. Sabe SA, Feng J, Sellke FW, Abid MR. Mechanisms and clinical implications of endothelium-dependent vasomotor dysfunction in coronary microvasculature. Am J Physiol Heart Circ Physiol. 2022;322:H819-41.
8. Borg R, Cherney DZ. Glomerular hyperfiltration and tubuloglomerular feedback in diabetic kidney disease: physiological insights and potential clinical translation. J Am Soc Nephrol. 2025;36:342-4.
9. Sharma N, Verma SK, Sharma S, Kapoor N, Kalra S. Diabetic platelets: pathophysiology, clinical significance, and therapeutic perspectives. Diabetes Ther. 2025;16:2101-9.
10. Iacobellis G, Bianco AC. Epicardial adipose tissue: emerging physiological, pathophysiological and clinical features. Trends Endocrinol Metab. 2011;22:450-7.
11. Vyas V, Blythe H, Wood EG, et al. Obesity and diabetes are major risk factors for epicardial adipose tissue inflammation. JCI Insight. 2021;6:e145495.
12. Zhou Y, Yan H, Zhu Y, Jiang W, Zhang S. Multifactorial mechanisms of obesity-related HFpEF: the central role of epicardial adipose tissue and therapeutic perspectives. Front Cardiovasc Med. 2025;12:1701459.
13. Al Qassab M, Chaarani N, Hamou A, et al. The gut microbiota-insulin resistance axis: mechanisms, clinical implications, and therapeutic potential. FASEB Bioadv. 2026;8:e70080.
14. Dong H, Sun Y, Nie L, et al. Metabolic memory: mechanisms and diseases. Signal Transduct Target Ther. 2024;9:38.
15. Loomba R, Hartman ML, Lawitz EJ, et al.; SYNERGY-NASH Investigators. Tirzepatide for metabolic dysfunction-associated steatohepatitis with liver fibrosis. N Engl J Med. 2024;391:299-310.
16. Akoumianakis I, Zagaliotis A, Konstantaraki M, Filippatos TD. GLP-1 analogs and regional adiposity: a systematic review and meta-analysis. Obes Rev. 2023;24:e13574.
17. Wang D, Morton JI, Magliano DJ, Shaw JE. Comparison of subcutaneous, visceral, liver and muscle fat depots in relation to prevalent and incident diabetes. Diabetes Obes Metab. 2025;27:6304-13.
18. Borel AL, Nazare JA, Smith J, et al. Visceral, subcutaneous abdominal adiposity and liver fat content distribution in normal glucose tolerance, impaired fasting glucose and/or impaired glucose tolerance. Int J Obes. 2015;39:495-501.
19. Kabahizi A, Wallace B, Lieu L, et al. Glucagon-like peptide-1 (GLP-1) signalling in the brain: from neural circuits and metabolism to therapeutics. Br J Pharmacol. 2022;179:600-24.
20. Lee YH, Lim S, Davies MJ. Cardiometabolic and renal benefits of sodium-glucose cotransporter 2 inhibitors. Nat Rev Endocrinol. 2025;21:783-98.
21. Marx N, Husain M, Lehrke M, Verma S, Sattar N. GLP-1 receptor agonists for the reduction of atherosclerotic cardiovascular risk in patients with type 2 diabetes. Circulation. 2022;146:1882-94.
22. McGuire DK, Marx N, Mulvagh SL, et al.; SOUL Study Group. Oral semaglutide and cardiovascular outcomes in high-risk type 2 diabetes. N Engl J Med. 2025;392:2001-12.
23. Moiz A, Filion KB, Tsoukas MA, Yu OHY, Peters TM, Eisenberg MJ. The expanding role of GLP-1 receptor agonists: a narrative review of current evidence and future directions. EClinicalMedicine. 2025;86:103363.
24. Lincoff AM, Brown-Frandsen K, Colhoun HM, et al.; SELECT Trial Investigators. Semaglutide and cardiovascular outcomes in obesity without diabetes. N Engl J Med. 2023;389:2221-32.
25. Deanfield J, Lincoff AM, Kahn SE, et al. Semaglutide and cardiovascular outcomes by baseline and changes in adiposity measurements: a prespecified analysis of the SELECT trial. Lancet. 2025;406:2257-68.
26. Nicholls SJ, Pavo I, Bhatt DL, et al.; SURPASS-CVOT Investigators. Cardiovascular outcomes with tirzepatide versus dulaglutide in type 2 diabetes. N Engl J Med. 2025;393:2409-20.
27. Wing RR, Bolin P, Brancati FL, et al.; Look AHEAD Research Group. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med. 2013;369:145-54.
28. Carbone S, Patel KV. Semaglutide in HFpEF and obesity: targeting both for functional gains. JACC Heart Fail. 2025;13:102665.
29. Kosiborod MN, Abildstrøm SZ, Borlaug BA, et al.; STEP-HFpEF Trial Committees and Investigators. Semaglutide in patients with heart failure with preserved ejection fraction and obesity. N Engl J Med. 2023;389:1069-84.
30. Borel AL, Nazare JA, Smith J, et al. Visceral and not subcutaneous abdominal adiposity reduction drives the benefits of a 1-year lifestyle modification program. Obesity. 2012;20:1223-33.
31. Ross R, Bradshaw AJ. The future of obesity reduction: beyond weight loss. Nat Rev Endocrinol. 2009;5:319-25.
32. Sattar N, Neeland IJ, Dahlqvist Leinhard O, et al. Tirzepatide and muscle composition changes in people with type 2 diabetes (SURPASS-3 MRI): a post-hoc analysis of a randomised, open-label, parallel-group, phase 3 trial. Lancet Diabetes Endocrinol. 2025;13:482-93.
33. Tuleta I, Frangogiannis NG. Fibrosis of the diabetic heart: clinical significance, molecular mechanisms, and therapeutic opportunities. Adv Drug Deliv Rev. 2021;176:113904.
34. Nidorf SM, Fiolet ATL, Mosterd A, et al.; LoDoCo2 Trial Investigators. Colchicine in patients with chronic coronary disease. N Engl J Med. 2020;383:1838-47.
35. Ridker PM, Everett BM, Thuren T, et al.; CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119-31.
36. Agarwal R, Filippatos G, Pitt B, et al.; FIDELIO-DKD and FIGARO-DKD investigators. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: the FIDELITY pooled analysis. Eur Heart J. 2022;43:474-84.
37. Rodriguez PJ, Zhang V, Gratzl S, et al. Discontinuation and reinitiation of dual-labeled GLP-1 receptor agonists among US adults with overweight or obesity. JAMA Netw Open. 2025;8:e2457349.
38. Alissou M, Demangeat T, Folope V, et al. Impact of Semaglutide on fat mass, lean mass and muscle function in patients with obesity: the SEMALEAN study. Diabetes Obes Metab. 2026;28:112-21.
39. Villareal DT, Aguirre L, Gurney AB, et al. Aerobic or resistance exercise, or both, in dieting obese older adults. N Engl J Med. 2017;376:1943-55.
40. Bauer J, Biolo G, Cederholm T, et al. Evidence-based recommendations for optimal dietary protein intake in older people: a position paper from the PROT-AGE Study Group. J Am Med Dir Assoc. 2013;14:542-59.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].